Přesné gen odstranění a nahrazení pomocí CRISPR/Cas9 systému v lidských buňkách
Zde jsme se ukázat, že dva průvodce RNAs spolu s Cas9 efektivně generovat DNA delece až 10 kb v lidských buňkách v procesu, kde oprava mazání je do značné míry dosaženo tím, přesné konci spojení. Kromě toho poskytujeme údaje, které ukazují, že systém CRISPR/Cas9 může nahradit velké genomické fragmenty za přítomnosti lineárního dárce homologní opravy.,
bakteriální seskupený pravidelně nahuštěny krátké opakování palindromické/ CRISPR—asociovaný (CRISPR/Cas) lokusy kódují RNA-řízené imunitní systémy, které chrání buňky proti invazi virů a plazmidů (1, 2). V Streptococcus pyogenes, typ II CRISPR/Cas systémy používají RNA-řízené endonukleázy (RGEN), Cas9, aby katalyzovat site-specifické štěpení cílové sekvence DNA., Zaměření Cas9 specifických genomových míst je zprostředkován 20 nukleotidů průvodce sekvence v rámci související CRISPR RNA (crRNA) a vyžaduje trans-aktivace crRNA (tracrRNA) verbuje crRNA do Cas9 komplexu (3). Rozpoznání štěpných míst je určeno párováním crrna-DNA báze a motivem sousedícím s protospacerem (PAM), sekvencí tří nukleotidů (NGG) vedle komplementární oblasti DNA (4)., Je pozoruhodné, že jeden guide RNA (gRNA), která napodobuje tracrRNA-crRNA komplex může zaměstnat Cas9 k cílené genomické stránky a vytvářet dvouvláknové zlomy (DSBs) v DNA (5). Systémy CRISPR / Cas9 byly přizpůsobeny pro editaci genomu specifického pro danou lokalitu v různých typech buněk a organismech (6-12).
editace genomu pomocí CRISPR / Cas9 je zahájena zavedením DSB na cílený genomický lokus pomocí RNA naprogramovaného RGEN. Následuje Oprava DSB buď homologně řízenou opravou (HDR), nebo nehomologním koncovým spojením (NHEJ)., Za přítomnosti homologního dárce opravy může být systém CRISPR/Cas9 použit k vytvoření přesných a definovaných úprav a vložení na cílený lokus prostřednictvím procesu HDR. Při absenci homologního dárce opravy jsou jednotlivé dsbs generované CRISPR/Cas9 opraveny pomocí nhej náchylného k chybám, což vede k mutacím vkládání nebo mazání (indel). Indelové mutace v kódujících exonech mohou zavádět předčasné Stop kodony nebo mutace frame-shift, čímž inaktivují odpovídající proteiny., Indel mutace generované z opravy jednoho DSB nemusí být užitečné experimenty zaměřené na charakterizaci funkční domény protein-kódujících genů nebo pro inaktivaci genomické prvky, jako jsou intergenic nebo intronic regulační sekvence nebo noncoding RNA genů. Odstranění fragmentů DNA v cílovém loci by poskytlo cestu ke studiu těchto funkčních prvků. Za tímto účelem bylo zavedeno více DSB pro generování delecí v Drosophile (12, 13), zebrafish (14) a lidských buňkách (8), i když s nízkou účinností., Cílené genomické DNA delece byly také dosaženo pomocí zinc finger nuclease (ZFN) nebo transcription activator-like effector nuclease (CHOVAT) v lidských buňkách (15-17). Účinnost těchto přístupů je však obecně nízká. Kromě toho zůstávají Zfns a TALENs poněkud obtížné a nákladné navrhnout, vyvinout a empiricky testovat v buněčném kontextu.
zde jsme zkoumali generování fragmentových delecí v lidských buňkách katalyzovaných systémem CRISPR / Cas9. Ukazujeme, že 2 gRNAs spolu s Cas9 mohou efektivně vytvářet delece DNA až 10 kb., Zajímavé je, že jsme zjistili, že oprava tohoto procesu mazání je do značné míry provedena přesným koncovým spojením. Kromě toho se zdá, že cílené vymazání pomocí CRISPR / Cas9 je nezávislé na transkripčním stavu cíleného lokusu. Nakonec ukážeme, že systém CRISPR/ Cas9 lze použít k nahrazení velkých genomických fragmentů za přítomnosti lineárního dárce homologní opravy.
Materiál a Metody
Plasmidu konstrukce
základní H1 promotor byl zesílen z pLVTHM plasmidu (Addgene, #12247, Cambridge, MA)., Oligonukleotidy obsahující modifikovaný promotor H1 a páteř požadovaných sekvencí gRNA se dvěma místy BsaI byly syntetizovány (PAN Facility, Stanfordská univerzita). Výsledné celovečerní produkty byly zesíleny PCR a klonovány do vektoru pUC19. Na ampicilin gen (amp) a H1 promotor ve vektoru pUC19 obsahují BsaI omezení enzym stránek; tyto byly mutované (amp gen byl změněn z G1601C, které nemění sekvenci aminokyselin; H1 promotor byl změněn z GAGACC na GAGGACC) k odstranění BsaI stránky., Protokol pro klonování gRNA je uveden v doplňkovém materiálu. Všechny sekvence cílových webů jsou uvedeny v doplňkové tabulce S1.
Buněčné kultury
HEK 293T, SK-Hep1, a HeLa buňky byly kultivovány v Dulbecco“s modified Eagle“s medium (DMEM) doplněném 10% fetálního bovinního séra (FBS) (Hyclone, Logan, UT) a penicilin/streptomycin (pen/strep) (Invitrogen, Carlsbad, CA). PC3 buňky byly kultivovány v prostředí RPMI-1640 doplněné o 10% FBS a pen/strep., Pro tumor nekrotizující faktor α (TNF-α) stimulace, 293T buňky byly ošetřeny s uvedenými koncentracemi TNF-α(R&D Systems, Minneapolis, MN). Buňky byly udržovány při 37 ° C a 5% CO2 ve zvlhčeném inkubátoru.
cílená delece DNA
HEK 293t buňky byly naočkovány do 12-dobře desek o hustotě 100.000 buněk na jamku. Po 24 h, buňky byly přechodně transfektovaly s 1 µg Cas9 plasmidu (Addgene, #41815), 0,5 µg gRNA T1, a 0,5 µg gRNA T2 plazmidů pomocí Lipofectamine 2000 (Invitrogen) podle výrobce“s protokoly., Genomická DNA byla extrahována 48 h po transfekci pomocí Quickextract dna Extraction Solution (Epicentre Biotechnologies, Madison, WI). Společná PCR byla provedena za účelem zesílení cílové oblasti pomocí primerů lemujících cílové oblasti. Divoké a zkrácené genomové fragmenty byly vyřešeny gelovou elektroforézou. Real-time PCR (RT-PCR) byla provedena kvantifikovat procento vymazání pomocí primery přes křižovatku nebo v rámci regionu mazání. Srovnávací metoda Cq byla použita pro výpočet úrovně exprese cílové oblasti vzhledem k referenční oblasti (actb lokus)., Procento delece v cílové oblasti bylo dále vypočítáno poměrem cílových buněk vzhledem k kontrolním buňkám. Všechny sekvence primerů jsou uvedeny v doplňkové tabulce S2.
Cíle sekvenování
Buňky byly sklizeny dva dny po transfekci, a genomová DNA byla extrahována pomocí QuickExtract DNA Extrakčního Roztoku (Epicentre Biotechnologies). PCR byla provedena s cílem zesílit cílení regionu s genomickou DNA pocházející z buněk, a amplikony byly hluboké seřazeny podle MiSeq Osobní Sekvencer (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Dárcovské sekvence jsou uvedeny v doplňkovém materiálu. Dvě 5 ‚ – Většina vazeb jsou fosforothioát (označený hvězdičkami). Buňky v 6-no desky byly přechodně transfektovaly s 2,0 µg Cas9 plasmidu, 0,8 µg gRNA T1 plasmidu, 0,8 µg gRNA T2 plasmidu, a 0,4 µg lineární dárce pomocí Lipofectamine 2000 (Invitrogen). Při 48 h po transfekci byly buňky ošetřeny 1 ng/mL TNF-α po dobu 24 h a poté byly tříděny GFP-pozitivní buňky.
luciferázy
Pro luciferase assay, HEK 293T buňky byly nasazeny do 96-no desek při hustotě 5000 buněk na jamku., Po 24 h, buňky byly přechodně transfektovaly s 5 ng pRL-TK Renilla luciferase reporter a 100 ng luciferase reporter s cytomegalovirus (CMV), SV40 (Simian virus 40), nebo základní pořadatel. Po 48 hodinách byla aktivita luciferázy měřena pomocí systému dvojitého luciferase reporter assay system (proma).
Western blot
proteiny byly odděleny dodecylsulfát sodný—PAGE (SDS-PAGE) a převedeny na nitrocelulózové membrány. Membrány byly blokovány 5% netučným mlékem a inkubovány s protilátkou GFP (CST, #2555S, Danvers, MA)., Komplex antigen-protilátka byl detekován pomocí zvýšených chemiluminiscenčních činidel.
výsledky a diskuse
upravili jsme bakteriální systém CRISPR/ Cas9 typu II pro mutagenizaci genomické DNA v lidských buňkách. Lidská kodon optimalizovaná verze proteinu s. pyogenes Cas9 s jaderným lokalizačním signálem C-terminus SV40 byla vyjádřena pomocí dříve popsaného systému (6). Pro přímé štěpení Cas9 na požadovanou sekvenci jsme vyjádřili crrna-tracrRNA fúzní přepisy, dále označované jako guide RNA (gRNAs), z modifikovaného lidského promotoru H1 polymerázy III., 3 ‚ konec promotoru H1 byl upraven tak, aby umožnil transkripci gRNAs, které začínají jakýmkoli nukleotidem. Omezena pouze na požadavek, aby 20 bp crRNA cíl být doplněno PAM sekvence, NGG (kde N je jakýkoli nukleotid), tento přístup může být v zásadě použit na cíl genomické umístění, které má formu N20NGG. Pro usnadnění klonování expresního vektoru gRNA jsme použili restrikční enzym typu IIs, BsaI. To vyžadovalo syntézu 24 bp oligonukleotidu obsahujícího oblast komplementarity na cílové místo na DNA., Jednoduchý a efektivní protokol pro klonování expresního vektoru gRNA (obrázek 1a) je podrobně popsán v doplňkovém materiálu.
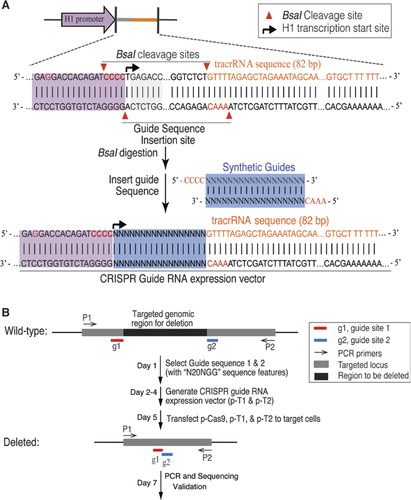
(A) návrh expresního vektoru vodicí RNA (gRNA). Vektor byl navržen tak, aby produkoval gRNA přepisy se syntetickou gRNA fúzovanou s trans-aktivační RNA / CRISPR RNA (tracrna)., Promotor H1 byl upraven tak, aby eliminoval vnitřní místo omezovacího enzymu BsaI typu IIS změnou gagacc v promotoru H1 na GAGGACC. A BsaI stránky byl představen vytvořit klonování stránek pro gRNA a tracrRNA fúze vložením syntetických oligonukleotidových duplexů s kompatibilní převisy. 3 ‚konec promotoru H1 byl upraven tak, aby byl CCACAGATCCCC, aby se usnadnila transkripce gRNA s jakýmkoli nukleotidem na konci 5‘. B) kroky cílené delece genů s CRISPR / Cas9.,
Pro smazání velká část genomové DNA, použili jsme pár gRNAs proti cílené lokusu (Obrázek 1B). Na hranici cílové oblasti byly vybrány dvě cílové lokality se vzorem N20NGG. Účinnost cíleného mazání vedená různými kombinacemi párů gRNA byla určena analýzami PCR pomocí primerů lemujících cílové oblasti. Divoké a zkrácené genomové fragmenty byly vyřešeny gelovou elektroforézou. Aby se zabránilo zkreslení zesílení PCR, bylo vymazání procenta kvantifikováno RT-PCR pomocí jednoho páru primerů., Primery byly navrženy přes deleční křižovatku (jeden primer mimo oblast delece, druhý primer v oblasti delece) nebo v oblastech delece (oba primery se nacházejí v oblasti delece). Tím je zesílen pouze jeden pás s párem primerů jak pro cílené buňky, tak pro kontrolní buňky. Jsme vypočítali procenta smazání porovnáním relativní množství produktů PCR (cílové buňky versus kontrolní buňky) zesílen stejný primer pair. Cílené odstranění bylo dále ověřeno sekvenováním.,
posoudit, jak gRNA páry by mohly mít vliv na následné opravy a generace delece, jsme se poprvé navržen sady gRNAs cílených proti lidské CDC42 genomické locus a odděleny vzdáleností v rozmezí přibližně od 200 do 10 000 bp (Obrázek 2A a Doplňková Tabulka S1). Poté jsme vyhodnotili schopnost každého páru gRNA generovat delece v lidských buňkách HEK 293T v přítomnosti Cas9. Robustní účinnost delecí založených na NHEJ (až 68%) byla potvrzena qPCR (obrázek 2b–2D)., Dokonce i pro vymazání genomické oblasti 10 kb jsme získali míru cílení 16% až 28%, v závislosti na páru gRNA. Tento proces editace zprostředkovaný RNA byl rychlý, přičemž první detekovatelná delece se objevila přibližně 12 h po transfekci (Doplňkový obrázek S1). Systém byl účinný u různých typů buněk, včetně: PC3, SK-Hep1 a HeLa buněk (Doplňkový obrázek S2).

(a) schematický diagram znázorňující umístění vodicích RNAs (gRNAs) zaměřených na lokus cdc42. (B–D) účinnost cílené delece s CRISPR/Cas9 byla stanovena PCR v buňkách HEK 293t. Byly použity primery mimo očekávané oblasti mazání. Procento vymazání bylo kvantifikováno RT-PCR pomocí primerů napříč křižovatkou nebo v oblasti mazání. E-F) účinnost a přesnost cíleného mazání v cdc42 byla potvrzena sekvenováním E) Sanger a F) sekvenačními analýzami s vysokou propustností. Produkt PCR obsahující pouze deleční amplicon byl obohacen pro sekvenování.,
Delece byly dále potvrzeny sekvenováním PCR produktů pokrývajících očekávané štěpení stránek. Sekvenování Sanger ukázalo deleční křižovatky vyplynuly z přesného ligace tupých DSBs vytvořených Cas9; každá DSB nastala přesně 3 bp před sekvencí PAM (obrázek 2E a doplňkový obrázek S3). Použili jsme také hluboké sekvenování smazání amplikony posoudit správnost vymazání účinnosti; asi 80% čte, cílené DSBs byly dokonale opraveny (Obrázek 2F).,
Jsme zrekapituloval tyto závěry testováním gRNA párů navržen tak, aby odstranit fragmenty z genomové lokusy obsahující mikrorna miR-21 genu v HEK 293T buňky. Dva gRNAs byly navrženy tak, aby se zaměřily na hranice miR – 21 vlásenky (Doplňkový obrázek S4). Účinnost delece byla 38% po transfekci dvěma gRNAs a Cas9, měřeno pomocí testu PCR (Doplňkový obrázek S4B). Hluboké sekvenování potvrdilo, že k vymazání došlo přesně podle očekávání (doplňující obrázek S4C).,
, Aby prošetřila, zda CRISPR/ Cas9-zprostředkované genové delece systém je ovlivněn transkripční stav cílené geny, gen kódující chemokinový (C-C motif) ligand 2 (CCL2) bylo cílené. CCL2 je malý cytokin patřící do rodiny CC chemokinů; Gen CCL2 je cílem signalizace NF-kB. Náhodně jsme vybrali osm cílových míst umístěných na koncích 5′ a 3′ genového lokusu CCL2 (obrázek 3a). Dosáhli jsme robustní a efektivní delece různých oblastí genu pomocí Cas9 a různých párů gRNAs v buňkách HEK 293t (obrázek 3B–3D).,

(a) schematický diagram znázorňující umístění vodicích RNAs (gRNAs) zaměřených na ccl2 lokus. (B–D) účinnost cílené delece CCL2 s CRISPR/Cas9 v buňkách HEK 293t. E) hladiny mRNA CCL2 byly stanoveny na základě přidání faktoru nekrózy nádorů α (TNF-α) kvantitativními analýzami RT-PCR v buňkách HEK 293t. Údaje byly zobrazeny s prostředky ± sem ve trojím vyhotovení., F) účinnost cílené delece CCL2 s CRISPR / Cas9 po ošetření TNF-α po dobu 24 hodin v buňkách HEK 293t. G) úroveň aktivity luciferázy z cytomegaloviru (CMV) nebo SV40 nebo základního promotoru v buňkách HEK 293t. Údaje byly zobrazeny s prostředky ± sem ve trojím vyhotovení. H) účinnost cílené delece genu luciferázy řízeného CMV nebo SV40 nebo základního promotoru v buňkách HEK 293t. (B–D,F,H) procento vymazání bylo kvantifikováno RT-PCR pomocí primerů přes křižovatku nebo v oblasti mazání.,
Exprese CCL2 gene byl dramaticky vyvolané TNF-α (k ∼300-násobné zvýšení) v HEK 293T buňky (Obrázek 3E), poskytuje dobrý model, aby prošetřila, zda CRISPR/ Cas9-zprostředkované cílené editování genomu je ovlivněna transkripční aktivitu. Zájmu, účinnost cílené vypuštění CCL2 gene locus nebyla ovlivněna tím, že ošetření buněk s TNF-α(Obrázek 3F), což naznačuje, že transkripce nemění CRISPR/Cas9-zprostředkované odstranění., K dalšímu potvrzení tohoto výsledku, jsme zaměřili exogenní reportér gen řízen různá předkladatelé s různými silné stránky, kde různé transkripční aktivity by mohly být hodnoceny pomocí luciferázy (Obrázek 3G). PCR testy odhalily podobné účinnosti cílené delece v HEK 293T buňky po ko-transfekce reporter gene společně s Cas9 a gRNA párů (Obrázek 3H). Tento výsledek naznačuje, že oprava zprostředkovaná NHEJ může nastat i přes výskyt různých stupňů transkripční aktivity.,
DSBs může stimulovat HDR, aby umožnil vysoce přesnou výměnu poškozené oblasti homologním dárcem. Abychom získali cílenou genomovou náhradu DNA, zavedli jsme do buněk pár gRNAs, Cas9 a lineárního dárce s homologií do cílové oblasti (obrázek 4a). Lineární dárce byl získán amplifikací PCR s primery nesoucími 50 bp homologní sekvence. Stejný dárce byl úspěšně vložen pomocí systému opravy HDR založeného na ZFN (18)., K testování proveditelnosti CRISPR/ Cas9-zprostředkovaná náhradní tím, HDR, zaměřili jsme se na CCL2 locus s párem gRNAs (#39 a #1854 je znázorněno na Obrázku 3) a dárce ložiskem enhanced zelený fluorescenční protein (EGFP) kódování sekvence a SV40 poly(A) stránky (Obrázek 4A; sekvence a pozice jsou uvedeny v Doplňující Materiál). Pomocí tohoto systému, přibližně 0,5% cílené buňky byly EGFP-pozitivní, vzhledem k tomu, že pouze materiálu 0,023% EGFP-pozitivní v mock transfekci buněk (jen transfektovaly s dárcem), který byl podobný kontrolní buňky (0.021%, bez transfekci)., EGFP-pozitivní buňky byly pak roztříděny průtokovou cytometrií. Site-specific integrace byla potvrzena PCR pomocí dvou párů primerů lemujících jak homologní zbraně, tak celý nahrazený region. Jak je znázorněno na Obrázku 4B, jsme pozorovali očekávané nahrazuje regionu obsahující plné délce sekvence EGFP a homologní zbraní (Sanger sekvenování výsledek je znázorněno na Doplňkový Materiál). Byla také zjištěna endogenní alela divokého typu (obrázek 4B), což naznačuje, že ne všechny alely jsou zaměřeny., Kromě toho jsme vybrali jeden klony z EGFP-pozitivní buňky, a zjistil, že všechny klony (6 z 6 zkoumaných) se očekává, že integrace (Obrázek 4C), ale endogenní wild-type alela byla také zjištěna ve třech klonů (Obrázek 4C), což naznačuje, že pouze jedna alela byla zaměřena v těchto klonů. Exprese EGFP bílkovin v cílových buňkách (EGFP-pozitivní buňky řazeny) byla up-regulována na TNF-α léčby jako hodnocena metodou Western blot a fluorescence activated cell sorting (FACS) (Obrázek 4, D a E)., Tyto výsledky ukázaly, že systém CRISPR/Cas9 lze použít k vytvoření genových/doménových náhrad s vysokou účinností a přesností.

(a) schematické diagramy znázorňující postup cílené genové náhrady za použití CRISPR / Cas9 v lidských buňkách., K testování účinnosti cílené nahrazování genů, průvodce RNAs (gRNAs) byly navrženy tak, aby odstranit uvedené oblasti (označené stránky 1 a 2) CCL2 gene a nahradit odstraněné oblasti s EGFP-polyA kazeta dárce se zbraněmi s krátkou oblastí homologie. Cílené stránky webu 1 a webu 2 jsou #39 a #1854 v genu CCL2 znázorněném na obrázku 3. Homologní sekvence (50 bp) jsou jen proti proudu a po proudu od míst mazání. Pozice a posloupnosti jsou podrobně uvedeny v doplňkovém materiálu., B) účinnost cílené genové náhrady CRISPR / Cas9 byla stanovena PCR v buňkách HEK 293t. Pro zesílení PCR byly použity primery překlenující křižovatky mezi CCL2 a EGFP. C) stanovení PCR pro cílenou genovou náhradu jednotlivých klonů. (D,E) exprese proteinu EGFP na přídavku TNF-α v buňkách HEK 293t byla určena D) Western blot a E) FACS.
zde popisujeme jednoduchý a účinný přístup k deleci genů pomocí systému CRISPR/Cas9., Jsme prokázali, že zavedení tohoto systému do lidských HEK 293T buňky, a jiné lidské typy buněk, indukované delece fragmentů až 10 kb s účinností v rozmezí mezi 11% a 68%, v závislosti na cílové sekvence. Schopnost efektivně a přesně odstranit genomové segmenty usnadní studium funkčních genomických prvků v lidských buňkách. Tento přístup může být potenciálně použit k cílení na jakýkoli genomický loci.
vyskytly se obavy ohledně specifičnosti systému CRISPR/Cas9 (19-21)., Chcete-li vyloučit nežádoucí fenotypy v důsledku mimocílových mutací, doporučujeme použít pro každou cílovou oblast alespoň dva různé páry gRNAs. V naší studii nebyla potřeba více párů gRNA hlavním omezením vzhledem k jednoduchosti a vysoké účinnosti tohoto systému. Je pozoruhodné, že různé páry gRNAs zaměřené na stejnou oblast pracovaly s vysokou účinností (obrázky 2 a 4). Dalším přístupem, jak se vyhnout nežádoucím mutacím, je použití metody double nickase (22, 23)., Také jsme úspěšně použili metodu double nickase ke generování delece genomické DNA, ale účinnost byla výrazně nižší.
je známo, že oprava DNA DSBs je do značné míry zprostředkované náchylné k chybám NHEJ, ve které oba konce jsou zpracovány a spojeny dohromady způsobem, který je často doprovázen nukleotidů inzerce a delece. Takové spojení náchylné k chybám bylo pozorováno při opravě DSBs vytvořených ZFNs nebo TALENs. Naproti tomu Oprava DSBs generovaná Cas9 a dvěma gRNAs byla velmi přesná., Naše výsledky naznačují, že přestávky jsou přímo ligovány bez konečného zpracování a odhalují dříve nedoceněnou výhodu dráhy NHEJ. Mechanismus, který vede k přesným ligacím, musí být stanoven. Jednou z možností je, že cílené vymazání pomocí Cas9 a dvou gRNAs vede ke křižovatce, která není rozpoznána ani jedním z původních gRNAs. Také jsme analyzovali efektivitu při generování indel mutací pro jednotlivé gRNA a gRNA dvojice (#39 a #224 gRNA na Obrázku 3A) pomocí Sangerova sekvenování PCR amplikony (TA klonování)., Zájmu, jsme pozorovali, že účinnost generování indel mutací pro jednotlivé gRNA byla poměrně nízká (9.5%, 2 z 21 klony #39 gRNA; 5%, 1 z 20 klonů pro #224 gRNA). Nicméně, gRNA pár generovány vysoké účinnosti indel mutace (50%, 10 z 20 klonů pro #39 a #224), který byl podobný testu pomocí qPCR (52%, Obrázek 3B). Navrhujeme, aby jedna gRNA často vyústila v jeden tupý konec štěpného místa, který bude přesně opraven NHEJEM. Účinnost generování mutace je tedy mnohem nižší při použití jedné gRNA než dvojice gRNAs.,
tato práce byla podpořena granty Národního zdravotního ústavu (č. DP1CA174421) a W. m. Keck foundation C.-Z. C a National Natural Science Foundation of China (č. 81101481) a Shanghai Medical Talent Training Program (č. XYQ2011048) na S. L. H. tento článek podléhá politice přístupu veřejnosti NIH.
konkurenční zájmy
autoři nehlásí žádné konkurenční zájmy.
doplňující údaje
Chcete-li zobrazit doplňující údaje, které doprovázejí tento dokument, navštivte webové stránky časopisu na adrese: www.,future-science.com/doi/suppl/10.2144/000114196