Præcis gen sletning og udskiftning ved hjælp af CRISPR/Cas9 system i menneskelige celler
Her viser vi, at to guide Rna kombineret med Cas9 effektivt at generere DNA, der er udgået af op til 10 kb i menneskelige celler i en proces, hvor reparation af sletningen er i vid udstrækning gennemført ved præcis ende sammenføjning. Derudover leverer vi data, der viser, at CRISPR / Cas9-systemet kan erstatte store genomiske fragmenter i nærværelse af en lineær homolog reparationsdonor.,
den bakterielle grupperet regelmæssigt interspaced korte palindromiske gentagelser/CRISPR—associerede (CRISPR / Cas) loci koder RNA-guidede immunsystemer, der beskytter celler mod invaderende vira og plasmider (1, 2). I Streptococcus pyogenes bruger type II CRISPR / Cas-systemer en RNA-styret endonuclease (rgen), Cas9, til at katalysere stedspecifik spaltning af mål-DNA-sekvenser., Målretning af Cas9 til specifikke genomisk steder er medieret af en 20 nukleotid guide sekvens i en tilhørende CRISPR RNA (crRNA) og kræver en trans-aktivering crRNA (tracrRNA), der rekrutterer de crRNA i Cas9 komplekse (3). Genkendelse af spaltningssteder bestemmes ved crRNA-DNA-baseparring og et protospacer-tilstødende motiv (PAM), en tre nukleotidsekvens (NGG) sidestillet med det DNA-komplementære område (4)., Det er bemærkelsesværdigt, at et enkelt guide RNA (gRNA), der efterligner tracrRNA-crRNA-komplekset, kan rekruttere Cas9 til målrettede genomiske steder og generere dobbeltstrengede pauser (DSB ‘ er) i DNA (5). CRISPR / Cas9-systemerne er blevet tilpasset til stedspecifik genomredigering i forskellige celletyper og organismer (6-12).
Genomredigering med CRISPR / Cas9 indledes med introduktionen af et DSB på et målrettet genomisk locus ved hjælp af RNA-programmeret RGEN. Dette efterfølges af reparationen af DSB gennem enten homology-directed repair (HDR) eller nonhomologous end-joining (NHEJ)., I nærvær af en homolog reparationsdonor kan CRISPR / Cas9-systemet anvendes til at generere præcise og definerede modifikationer og Indsætninger på et målrettet locus gennem HDR-processen. I mangel af en homolog reparation donor, enkelt DSBs genereret af CRISPR/Cas9 er repareret gennem den fejlbehæftede NHEJ, hvilket resulterer i en insertion eller deletion (indel) mutationer. Indel-mutationer i kodende eksoner kan indføre for tidlige stopkodoner eller frame-shift-mutationer, hvorved de tilsvarende proteiner inaktiveres., Indel-mutationer genereret ved reparation af en enkelt DSB er muligvis ikke nyttige i forsøg, der sigter mod at karakterisere de funktionelle domæner for proteinkodende gener eller til inaktivering af genomiske elementer, såsom intergeniske eller introniske regulatoriske sekvenser eller ikke-kodende RNA-gener. DNA fragment sletninger i target loci ville give en vej til at studere disse funktionelle elementer. Til dette formål er der indført flere DSB ‘ er for at generere sletninger i Drosophila (12, 13), 14ebrafisk (14) og humane celler (8), omend med lav effektivitet., Målrettede genomiske DNA-deletioner er også opnået ved anvendelse af fingerinkfingernuklease (.fn) eller transkriptionsaktivatorlignende effektor nuklease (TALEN) i humane celler (15-17). Effektiviteten af disse tilgange er imidlertid generelt lav. Derudover forbliver ZFN ‘ er og TALENs noget vanskelige og dyre at designe, udvikle og empirisk teste i den cellulære kontekst.
Her undersøgte vi genereringen af fragment deletioner i humane celler katalyseret af CRISPR / Cas9-systemet. Vi viser, at 2 grna ‘ er kombineret med Cas9 effektivt kan oprette DNA-sletninger på op til 10 kb., Af interesse fandt vi, at reparation af denne sletningsproces stort set opnås ved præcis slutforbindelse. Desuden synes målrettet sletning med CRISPR / Cas9 at være uafhængig af den transkriptionelle status for det målrettede locus. Endelig viser vi, at CRISPR/ Cas9-systemet kan bruges til at erstatte store genomiske fragmenter i nærværelse af en lineær homolog reparationsdonor.
Materiale og Metoder
Plasmid konstruktion
De grundlæggende H1 middel blev forstærket af pLVTHM plasmid (Addgene, #12247, Cambridge, MA)., Oligonukleotider indeholdende den modificerede H1-promotor og rygrad af ønskede gRNA-sekvenser med to BsaI-steder blev syntetiseret (PAN Facility, Stanford University). De resulterende produkter i fuld længde blev forstærket ved PCR og klonet ind i pUC19-vektoren. Den ampicillin gen (amp) og H1 promotor i pUC19 vektor indeholder BsaI restriktionsenzym steder; disse var muteret (amp-gen blev ændret fra G1601C, der ikke ændrer aminosyresekvensen; H1 promotor blev ændret fra GAGACC at GAGGACC) til at fjerne BsaI sites., Protokollen for grna kloning er præsenteret i det supplerende materiale. Alle målretning sites sekvenser er præsenteret i supplerende tabel S1.
Celle kultur
HEK 293T, SK-Hep1, og HeLa celler, der blev dyrket i Dulbecco”s modificerede Ørnen”s medium (DMEM) suppleret med 10% føtalt bovint serum (FBS) (Hyclone, Logan, UT) og penicillin/streptomycin (pen/strep) (Invitrogen, Carlsbad, CA). PC3-celler blev dyrket i RPMI-1640 medium suppleret med 10% FBS og pen/strep., Til stimulering af tumornekrosefaktor α (TNF-α) blev 293T-celler behandlet med indikerede koncentrationer af TNF-α(r&D-systemer, Minneapolis, MN). Cellerne blev opretholdt ved 37 and C og 5% CO2 i en befugtet inkubator.
målrettet DNA-deletion
HEK 293T-celler blev podet i 12-brøndsplader med en densitet på 100.000 celler pr. Efter 24 h, cellerne var forbigående transfected med 1 µg Cas9 plasmid (Addgene, #41815), 0.5 µg gRNA T1, og 0,5 µg gRNA T2 plasmider hjælp Lipofectamine 2000 (Invitrogen) i henhold til producenten”s protokoller., Genomisk DNA blev ekstraheret 48 timer efter transfektion ved anvendelse af DNAUICKE .tract DNA-Ekstraktionsopløsning (Epicenter Biotechnologies, Madison, ,i). Fælles PCR blev udført for at forstærke den målrettede region ved hjælp af primere, der flankerer de målrettede regioner. Vildtype og trunkerede genomiske fragmenter blev løst ved gelelektroforese. Real-time PCR (RT-PCR) blev udført for at kvantificere procentdelen af deletion ved hjælp af primere på tværs af krydset eller inden for deletionsområdet. Den komparative C. – metode blev brugt til at beregne ekspressionsniveauet for målregionen i forhold til en referenceregion (ACTB locus)., Procent af sletning i målområdet blev yderligere beregnet ved forholdet mellem målceller i forhold til kontrolceller. Alle grundingssekvenser er angivet i supplerende tabel S2.
Målsekvensering
celler blev høstet to dage efter transfektion, og det genomiske DNA blev ekstraheret ved hjælp af DNAUICKE .tract DNA-Ekstraktionsopløsning (Epicenter Biotechnologies). PCR blev udført for at forstærke målområdet med genomisk DNA afledt af cellerne, og ampliconer blev dybt sekventeret af mise.Personal se .uencer (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Donorsekvenserne præsenteres i det supplerende materiale. De to 5′-mest forbindelser er phosphorothioat (angivet med stjerner). Celler i 6-brønds plader blev transient transficeret med 2,0 µg Cas9 plasmid, 0,8 µg gRNA T1 plasmid, 0,8 µg gRNA T2 plasmid og 0,4 µg lineær donor under anvendelse af Lipofectamine 2000 (Invitrogen). Ved 48 timer efter transfektion blev celler behandlet med 1 ng / mL TNF-α i 24 timer, og derefter blev GFP-positive celler sorteret.
Luciferase assay
for luciferase assayet blev HEK 293T celler podet i 96-brønds plader ved en densitet på 5000 celler pr., Efter 24 h, cellerne var forbigående transfected med 5 ng af pRL-TK Renilla luciferase reporter og 100 ng af luciferase reporter med cytomegalovirus (CMV), SV40 (Simian virus 40), eller grundlæggende promotor. Efter 48 timer blev luciferaseaktiviteten målt med det dobbelte luciferase reporter assay system (Promega).
Westernestern blot
proteiner blev adskilt af natriumdodecylsulfat—side (SDS-side) og overført til nitrocellulosemembraner. Membranerne blev blokeret med 5% fedtfri mælk og inkuberet med GFP-antistof (CST, #2555S, Danvers, MA)., Antigen-antistofkomplekset blev påvist med forbedrede kemiluminescensreagenser.
resultater og diskussion
vi tilpassede bakterietype II CRISPR / Cas9-systemet til mutageni .e genomisk DNA i humane celler. Den humane kodonoptimerede version af S. pyogenes Cas9-protein, der bærer et C-terminus SV40-kernelokaliseringssignal, blev udtrykt ved anvendelse af et tidligere beskrevet system (6). Direkte Cas9 spaltning til den ønskede sekvens, som vi gav udtryk for crRNA-tracrRNA fusion afskrifter, herefter henvist til som guide Rna (gRNAs), fra en modificeret menneskelige H1-polymerase III promotor., 3 ‘- enden af H1-promotoren blev ændret for at tillade transkription af grna ‘ er, der begynder med ethvert nukleotid. Begrænset kun af kravet om, at 20 bp crRNA-målet følges af PAM-sekvensen, NGG (hvor n er ethvert nukleotid), kan denne tilgang i princippet bruges til at målrette enhver genomisk placering, der har formen N20NGG. For at lette kloningen af gRNA-ekspressionsvektoren brugte vi et type IIS-restriktionsen .ym, bsai. Dette krævede syntese af et 24 bp-oligonukleotid indeholdende et komplementaritetsområde med målstedet på DNA ‘ et., Den enkle og effektive protokol til kloning af gRNA-ekspressionsvektoren (figur 1A) er beskrevet detaljeret i det supplerende materiale.
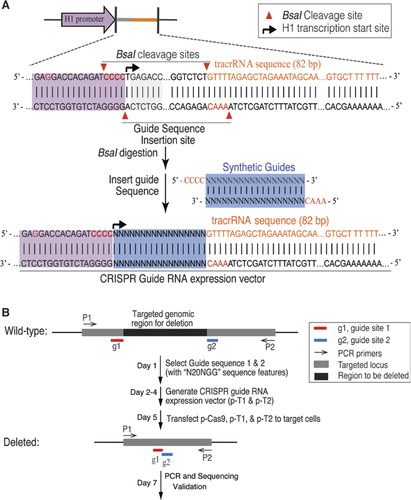
(A) Design af guide RNA (gRNA) ekspressionsvektor. Vektoren blev designet til at producere gRNA-transkripter med et syntetisk gRNA smeltet sammen med det transaktiverende RNA/CRISPR RNA (tracrRNA)., H1 promotor blev ændret for at fjerne den interne type IIS restriktionsenzym BsaI websted ved at ændre GAGACC i H1 initiativtager til GAGGACC. En BsaI stedet blev indført for at skabe kloning sites for gRNA og tracrRNA fusioner ved indsættelse af syntetiske oligonukleotid dobbelthuse med kompatible udhæng. 3 ‘- enden af H1-promotoren blev ændret til CCACAGATCCCC for at lette transkription af grna’ er med ethvert nukleotid i 5 ‘ – enden. (B) trinnene til målrettet genudsletning med CRISPR/Cas9.,
til sletning af et stort segment af genomisk DNA brugte vi et par grna ‘ er mod det målrettede locus (figur 1B). To målsteder med mønsteret N20NGG blev valgt ved grænsen for målområdet. Effektiviteten af målrettet sletning styret af forskellige kombinationer af gRNA-par blev bestemt ved PCR-analyser ved hjælp af primere, der flankerer de målrettede regioner. Vildtype og trunkerede genomiske fragmenter blev løst ved gelelektroforese. For at undgå PCR-amplifikationsbias blev procent deletion kvantificeret ved RT-PCR ved anvendelse af et primerpar., Primerne blev designet på tværs af sletningskrydset (en primer uden for sletningsområdet, den anden primer inden for sletningsområdet) eller inden for sletningsområderne (begge primere finder inden for sletningsområdet). Således forstærkes kun et enkelt bånd med primerparet for både de målrettede celler og kontrolceller. Vi beregnet procent deletion ved at sammenligne den relative mængde PCR-produkter (målceller versus kontrolceller) forstærket af det samme primerpar. Målrettede sletninger blev yderligere verificeret ved sekventering.,
for At vurdere, hvordan gRNA par kan påvirke de efterfølgende reparation og produktion af sletninger, vi først konstrueret sæt af gRNAs målrettet mod den menneskelige CDC42 genomisk locus og adskilt af afstande, der spænder fra cirka 200 til 10.000 bp (Figur 2A og Supplerende skema S1). Vi vurderede derefter hvert gRNA-pars evne til at generere sletninger i humane HEK 293T-celler i nærværelse af Cas9. Robust effektivitet af NHEJ-baserede sletninger (op til 68%) blev bekræftet af qPCR (figur 2B–2D)., Selv for sletning af en 10 kb genomisk region opnåede vi målretningshastigheder på 16% til 28% afhængigt af gRNA-parret. Denne RNA-medierede redigeringsproces var hurtig, hvor den første påviselige deletion forekom cirka 12 timer efter transfektion (supplerende figur S1). Systemet var effektivt i en række celletyper, herunder: PC3, SK-Hep1 og HeLa celler (supplerende figur S2).

(a) skematisk diagram, der viser placeringen af guide RNA ‘er (grna’ er) rettet mod CDC42 locus. (B–D) effektiviteten af målrettede sletning med CRISPR/Cas9 blev bestemt ved PCR i HEK 293T celler. Primere uden for forventede sletningsregioner blev anvendt. Procentdelen af sletningen blev kvantificeret ved RT-PCR ved hjælp af primere på tværs af krydset eller inden for sletningsområdet. (E–f) effektiviteten og præcisionen af målrettet sletning i CDC42 blev bekræftet af (e) Sanger sekventering og (f) high-throughput sekventering analyser. PCR-produktet, der kun indeholdt deletionampliconen, blev beriget til sekventering.,
sletninger blev yderligere bekræftet ved sekventering af PCR-produkter, der spænder over de forventede spaltningssteder. Sanger-sekventering viste sletning kryds resulteret i præcis ligering af den stumpe-endte DSBs skabt af Cas9; hver DSB fandt sted præcis 3 bp opstrøms PAM sekvens (Figur 2E og Supplerende Figur S3). Vi brugte også dyb sekventering af sletningsampliconer til at vurdere nøjagtigheden af sletningseffektiviteten; i omkring 80% af læsningerne blev de målrettede DSB ‘ er perfekt repareret (figur 2F).,
Vi rekapitulerede disse fund ved at teste gRNA-par designet til at slette fragmenter fra et genomisk locus indeholdende microRNA miR-21-genet i HEK 293T-celler. To grna ‘ er blev designet til at målrette grænserne for mir-21 hårnålen (supplerende figur S4). Deletionseffektiviteten var 38% efter transfektion med de to grna ‘ er og Cas9, målt ved hjælp af en PCR-assay (supplerende figur S4B). Deep se .uencing bekræftede, at sletningen skete nøjagtigt som forventet (supplerende figur S4C).,
for at undersøge, om det CRISPR / Cas9-medierede gen-deletionssystem påvirkes af transkriptionstilstanden for målrettede gener, blev genet, der koder for kemokinen (C-C-motiv) ligand 2 (CCL2), målrettet. CCL2 er et lille cytokin, der tilhører CC-kemokinfamilien; CCL2-genet er et mål for NF-kB-signalering. Vi valgte tilfældigt otte målsteder placeret i 5′ og 3 ‘ enderne af CCL2-genlokuset (figur 3A). Vi opnåede robust og effektiv sletning af forskellige regioner af genet ved hjælp af Cas9 og forskellige par grna ‘ er i HEK 293T–celler (figur 3B-3D).,

(a) skematisk diagram, der viser placeringen af guide RNA ‘er (grna’ er) rettet mod CCL2 locus. (B–D) effektiviteten af målrettede sletning af CCL2 med CRISPR/Cas9 i HEK 293T celler. (E) CCL2-mRNA-niveauer blev bestemt ved tumornekrosefaktor addition (TNF-α) – tilsætning ved kvantitative RT-PCR-analyser i HEK 293T-celler. Data blev vist med midlerne ± sem i tre eksemplarer., (F) effektiviteten af målrettede sletning af CCL2 med CRISPR/Cas9 efter behandling med TNF-α i 24 timer i HEK 293T celler. (G) niveauet af luciferase-aktivitet fra cytomegalovirus (CMV), eller SV40, eller en grundlæggende promotor i HEK 293T celler. Data blev vist med midlerne ± sem i tre eksemplarer. H) effektiviteten af målrettet deletion af et luciferase-gen kontrolleret af CMV eller SV40 eller en basispromotor i HEK 293T-celler. (B-D,F, H) procentdelen af sletningen blev kvantificeret ved RT-PCR ved hjælp af primere på tværs af krydset eller inden for sletningsområdet.,
Udtryk for CCL2 gen blev voldsomt provokeret af TNF-α (op til ∼300-fold stigning i HEK 293T celler (Figur 3E), hvilket giver en god model til at undersøge, om den CRISPR/ Cas9-medieret målrettet genom-redigering er påvirket af transkriptionel aktivitet. Af interesse blev effektiviteten af målrettet deletion af CCL2-genlokuset ikke påvirket af behandling af celler med TNF-α(figur 3F), hvilket antydede, at transkription ikke ændrede CRISPR/Cas9-medieret deletion., For yderligere at bekræfte dette resultat målrettede vi et eksogent reportergen drevet af forskellige promotorer med forskellige styrker, hvor de forskellige transkriptionelle aktiviteter kunne evalueres ved hjælp af en luciferase-assay (figur 3G). PCR-analyserne afslørede tilsvarende effektivitet i målrettede sletninger i HEK 293T celler efter co-transfektion af reporter gen sammen med Cas9 og gRNA par (Figur 3H). Dette resultat indikerer, at NHEJ-medieret reparation kan forekomme på trods af forekomsten af forskellige grader af transkriptionel aktivitet.,
DSB ‘ er kan stimulere HDR til at muliggøre meget præcis udskiftning af det beskadigede område med en homolog donor. For at opnå målrettet genomisk DNA-udskiftning introducerede vi et par grna ‘ er, Cas9 og en lineær donor med homologi til den målrettede region i celler (figur 4A). Den lineære donor blev opnået ved PCR-amplifikation med primere med en 50 bp homolog sekvens. Den samme donor blev med succes indsat ved hjælp af et repairfn-baseret HDR-reparationssystem (18)., For at teste muligheden for at CRISPR/ Cas9-medieret udskiftning af HDR, vi målrettet CCL2 locus med et par gRNAs (#39 og #1854 vist i Figur 3) og en donor, der bærer forbedret grønne fluorescerende protein (EGFP) kodende sekvens og SV40 poly(A) ejendommen (Figur 4A; sekvenser og positioner, som er præsenteret i det Supplerende Materiale). Ved hjælp af dette system, cirka 0,5% af målrettede celler var EGFP-positive, mens kun 0.023% var EGFP-positive i mock transfektion af celler (bare transfected med donor), som var magen til kontrol celler (0.021%, uden transfektion)., De EGFP-positive celler blev derefter sorteret efter Flo .cytometri. Stedspecifik integration blev bekræftet ved PCR ved hjælp af to par primere, der flankerer både de homologe arme og hele det udskiftede område. Som vist i figur 4B observerede vi det forventede udskiftede område, der indeholdt EGFP-sekvensen i fuld længde og de homologe arme (Sanger-sekventeringsresultat vist i det supplerende materiale). Den endogene allel af vildtype blev også påvist (figur 4B), hvilket indikerer, at ikke alle alleler er målrettet., Vi har endvidere valgt et enkelt kloner fra EGFP-positive celler og konstateret, at alle de kloner (6 af 6 undersøgt) havde den forventede integration (Figur 4C), men den endogene wild-type-allel, også blev fundet i tre af de kloner (Figur 4C), hvilket tyder på, at kun én allel var målrettet i disse kloner. Ekspression af EGFP-protein i målrettede celler (EGFP-positive sorterede celler) blev opreguleret ved TNF-treatment-behandling som evalueret ved fluoresestern blot og fluorescensaktiveret cellesortering (FACS) (figur 4, D og E)., Disse resultater viste, at CRISPR / Cas9-systemet kan bruges til at skabe gen – /domæneudskiftninger med høj effektivitet og nøjagtighed.

(a) skematiske diagrammer, der viser proceduren for målrettet genudskiftning ved anvendelse af CRISPR/Cas9 i humane celler., For at teste effekten af målrettet gen udskiftning, guide Rna (gRNAs) var designet til at slette det angivne område (markeret med lokaliteter 1 og 2) af den CCL2 gen og til at erstatte de slettede region med EGFP-polyA kassette donor med armene, med korte regioner af homologi. De målrettede steder af sted 1 og sted 2 er #39 og # 1854 inden for CCL2 genet vist i figur 3. De homologe sekvenser (50 bp) er lige opstrøms og nedstrøms for deletionsstederne. Positionerne og sekvenserne i detaljer er præsenteret i det supplerende materiale., (B) effektiviteten af målrettet gen udskiftning med CRISPR/Cas9 blev bestemt ved PCR i HEK 293T celler. Primere, der spænder over forbindelserne mellem CCL2 og EGFP, blev anvendt til PCR-amplifikation. C) PCR-assay til målrettet genudskiftning af de enkelte kloner. (D,e) ekspression af EGFP-protein ved TNF-addition-tilsætning i HEK 293T-celler blev bestemt ved (d) Westernestern blot og (e) FACS.
Her beskriver vi en enkel og effektiv tilgang til gen-sletning ved hjælp af den CRISPR/Cas9 system., Vi viste, at indførelsen af dette system i menneskers HEK 293T celler, og andre humane celletyper, der fremkaldes, der er udgået af fragmenter op til 10 kb med effektivitetsgevinster på mellem 11% og 68%, afhængigt af den målrettede forløb. Evnen til effektivt og præcist at slette genomiske segmenter vil lette undersøgelsen af funktionelle genomiske elementer i humane celler. Denne tilgang kan potentielt bruges til at målrette enhver genomisk loci.
der har været bekymring for specificiteten af CRISPR / Cas9-systemet (19-21)., For at udelukke uønskede fænotyper på grund af mutationer uden for målet foreslår vi, at der anvendes mindst to forskellige par grna ‘ er for hvert Målområde. I vores undersøgelse, behovet for flere gRNA-par var ikke en stor begrænsning, i betragtning af enkelheden og meget effektiviteten af dette system. Det er bemærkelsesværdigt, at forskellige par grna ‘ er målrettet mod samme region arbejdede med høj effektivitet (figur 2 og 4). En anden tilgang til at undgå uønskede mutationer er brugen af dobbelt nickase-metoden (22, 23)., Vi har også med succes anvendt den dobbelte nickase metode til at generere sletning af genomisk DNA, men effektiviteten var betydeligt lavere.
det er kendt, at reparationen af DNA DSB ‘ er i vid udstrækning formidles af fejlbehæftet NHEJ, hvor de to ender behandles og ligeres sammen på en måde, der ofte ledsages af nukleotidindsættelser og sletninger. En sådan fejl-tilbøjelige ende sammenføjning blev observeret i reparation af DSBs skabt af orfns eller TALENs. I modsætning hertil var reparationen af DSB ‘er genereret af Cas9 og to grna’ er meget præcis., Vores resultater antyder, at pauserne er direkte ligeret uden slutbehandling, hvilket afslører en tidligere ikke værdsat fordel ved NHEJ-vejen. Mekanismen, der resulterer i de præcise ligationer, skal stadig bestemmes. En mulighed er, at målrettet sletning ved hjælp af Cas9 og to grna ‘er resulterer i et kryds, der ikke genkendes af nogen af de originale grna’ er. Vi har også analyseret effektiviteten i at generere indel mutationer for den enkelte gRNA og gRNA par (#39 og #224 gRNA i Figur 3A) af Sanger-sekventering af PCR-amplifikation (TA-kloning)., Af interesse, vi har observeret, at effektiviteten af generere indel mutationer for enkelt gRNA var ret lav (9.5%, 2 af 21 kloner til #39 gRNA; 5%, 1 af 20 kloner til #224 gRNA). Imidlertid genererede gRNA-parret høj effektivitet af indel-mutationer (50%, 10 af 20 kloner for #39 og #224), hvilket svarede til assayet under anvendelse afppcr (52%, figur 3B). Vi foreslår, at en enkelt gRNA ofte resulterer i en stump ende af spaltningsstedet, som vil blive præcist repareret af NHEJ. Således er effektiviteten ved at generere mutation meget lavere ved brug af et enkelt gRNA end et par grna ‘ er.,
anerkendelser
dette arbejde blev støttet af tilskud fra National Institute of Health (nr. DP1CA174421) og W. M. Keck foundation-c-Z, C, og National Natural Science Foundation of China (Nr 81101481) og Shanghai Medical Talent Training Program (Nr. Thisy .2011048) til S. L. H. dette papir er underlagt NIH offentlig adgang politik.
konkurrerende interesser
forfatterne erklærer ingen konkurrerende interesser.
supplerende data
for at se de supplerende data, der følger med dette papir, kan du besøge tidsskriftets websiteebsted på: W…,future-science.com/doi/suppl/10.2144/000114196