Präzise Gen-Deletion und-ersatz unter Verwendung des CRISPR / Cas9-Systems in menschlichen Zellen
Hier zeigen wir, dass zwei mit Cas9 gekoppelte Leitschnas effizient DNA-Deletionen von bis zu 10 kb in menschlichen Zellen erzeugen in einem Prozess, bei dem die Reparatur der Deletion weitgehend durch präzise Endverbindung erreicht wird. Darüber hinaus liefern wir Daten, die zeigen, dass das CRISPR/Cas9-System große genomische Fragmente in Gegenwart eines linearen homologen Reparaturspenders ersetzen kann.,
Die Bakterien gruppierten regelmäßig interspaced short palindromic repeats / CRISPR-associated (CRISPR/Cas) loci kodieren RNA-geführte Immunsysteme, die Zellen vor eindringenden Viren und Plasmiden schützen (1, 2). In Streptococcus pyogenes verwenden die CRISPR/Cas-Systeme vom Typ II eine RNA-geführte Endonuklease (RGEN), Cas9, um ortsspezifische Spaltung von Ziel-DNA-Sequenzen zu katalysieren., Das Targeting von Cas9 auf bestimmte genomische Stellen wird durch eine 20-Nukleotid-Leitsequenz innerhalb einer assoziierten CRISPR-RNA (crRNA) vermittelt und erfordert eine transaktivierende crRNA (tracrRNA), die die crRNA in den Cas9-Komplex rekrutiert (3). Die Erkennung von Spaltstellen wird durch crRNA-DNA-Basenpaarung und ein Protospacer-benachbartes Motiv (PAM), einer der DNA-Komplementärregion (4) gegenüberliegenden Drei Nukleotidsequenz (NGG), bestimmt., Es ist bemerkenswert, dass eine einzelne leitende RNA (gRNA), die den tracrRNA-crRNA-Komplex nachahmt, Cas9 an gezielte genomische Stellen rekrutieren und doppelsträngige Brüche (DSBs) in DNA erzeugen kann (5). Die CRISPR / Cas9-Systeme wurden für die ortsspezifische Genombearbeitung in verschiedenen Zelltypen und Organismen angepasst (6-12).
Die Genombearbeitung mit CRISPR / Cas9 wird mit der Einführung eines DSB an einem gezielten genomischen Ort unter Verwendung des RNA-programmierten RGEN eingeleitet. Danach erfolgt die Reparatur des DSB entweder durch homologiegenerichtete Reparatur (HDR) oder nichthomologe Endverbindung (NHEJ)., In Gegenwart eines homologen Reparaturspenders kann das CRISPR / Cas9-System verwendet werden, um präzise und definierte Modifikationen und Einfügungen an einem Zielort durch den HDR-Prozess zu erzeugen. In Abwesenheit eines homologen Reparaturspenders werden einzelne von CRISPR/Cas9 erzeugte DSBs durch das fehleranfällige NHEJ repariert, was zu Insertions-oder Deletionsmutationen (Indel) führt. Indel-Mutationen in kodierenden Exonen können vorzeitige Stop-Codons oder Frame-Shift-Mutationen einführen, wodurch die entsprechenden Proteine inaktiviert werden., Indel-Mutationen, die durch die Reparatur eines einzelnen DSB erzeugt werden, können in Experimenten zur Charakterisierung der funktionellen Domänen proteinkodierender Gene oder zur Inaktivierung genomischer Elemente wie intergener oder intronischer regulatorischer Sequenzen oder nichtkodierender RNA-Gene nicht nützlich sein. DNA-Fragmentlöschungen in Zielloci würden einen Weg bieten, diese funktionellen Elemente zu untersuchen. Zu diesem Zweck wurden mehrere DSBs eingeführt, um Deletionen in Drosophila (12, 13), Zebrafischen (14) und menschlichen Zellen (8) zu erzeugen, wenn auch mit geringer Effizienz., Gezielte genomische DNA-Deletionen wurden auch mit Zinkfingernuklease (ZFN) oder transkriptionsaktivatorähnlicher Effektornuclease (TALEN) in menschlichen Zellen (15-17) erreicht. Die Effizienz dieser Ansätze ist jedoch im Allgemeinen gering. Darüber hinaus bleiben ZFNs und TALENs im zellulären Kontext etwas schwierig und teuer zu entwerfen, zu entwickeln und empirisch zu testen.
Hier untersuchten wir die Erzeugung von Fragmentlöschungen in menschlichen Zellen, die durch das CRISPR/Cas9-System katalysiert wurden. Wir zeigen, dass 2 gRNAs in Verbindung mit Cas9 DNA-Löschungen von bis zu 10 kb effizient erzeugen können., Von Interesse, Wir fanden heraus, dass die Reparatur dieses Löschvorgangs weitgehend durch präzise Endverbindung erreicht wird. Darüber hinaus scheint eine gezielte Löschung mit CRISPR/Cas9 unabhängig vom Transkriptionsstatus des Zielortes zu sein. Schließlich zeigen wir, dass das CRISPR / Cas9-System verwendet werden kann, um große genomische Fragmente in Gegenwart eines linearen homologen Reparaturspenders zu ersetzen.
Material und Methoden
Plasmidkonstruktion
Der grundlegende H1-Promotor wurde aus pLVTHM-Plasmid verstärkt (Addgene, #12247, Cambridge, MA)., Oligonukleotide, die den modifizierten H1-Promotor und das Rückgrat der gewünschten gRNA-Sequenzen mit zwei BsaI-Stellen enthielten, wurden synthetisiert (PAN-Anlage, Stanford University). Die resultierenden Produkte in voller Länge wurden durch PCR amplifiziert und in den pUC19-Vektor geklont. Das Ampicillin-Gen (amp) und der H1-Promotor im pUC19-Vektor enthalten BsaI-Restriktionsenzymstellen; diese wurden mutiert (das Amp-Gen wurde von G1601C geändert, was die Aminosäuresequenz nicht verändert; der H1-Promotor wurde von GAGACC zu GAGGACC geändert), um die BsaI-Stellen zu eliminieren., Das Protokoll für das gRNA-Klonen ist im ergänzenden Material dargestellt. Alle Targeting-Sites-Sequenzen sind in der ergänzenden Tabelle S1 dargestellt.
Zellkultur
HEK 293T, SK-Hep1 und HeLa-Zellen wurden in Dulbecco ’s modifiziertem Eagle‘ s Medium (DMEM) kultiviert, ergänzt mit 10% fetalem Rinderserum (FBS) (Hyklon, Logan, UT) und Penicillin/Streptomycin (Pen / strep) (Invitrogen, Carlsbad, CA). PC3-Zellen wurden in RPMI-1640-Medium kultiviert, ergänzt mit 10% FBS und Pen/Strep., Zur Stimulierung des Tumornekrosefaktors α (TNF-α) wurden 293T-Zellen mit indizierten Konzentrationen von TNF-α behandelt(R&D Systems, Minneapolis, MN). Die Zellen wurden bei 37°C und 5% CO2 in einem befeuchteten Inkubator gehalten.
Gezielte DNA-Deletion
HEK 293T-Zellen wurden in 12-Well-Platten mit einer Dichte von 100.000 Zellen pro Well ausgesät. Nach 24 h wurden die Zellen gemäß den Protokollen des Herstellers vorübergehend mit 1 µg Cas9-Plasmid (Addgen, #41815), 0,5 µg gRNA T1 und 0,5 µg gRNA T2-Plasmiden unter Verwendung von Lipofectamin 2000 (Invitrogen) transfektiert., Genomische DNA wurde 48 h nach der Transfektion mit QuickExtract DNA Extraction Solution extrahiert (Epicentre Biotechnologies, Madison, WI). Gemeinsame PCR wurde durchgeführt, um die Zielregion mit Primern zu verstärken, die die Zielregionen flankieren. Wildtyp – und abgeschnittene Genomfragmente wurden durch Gelelektrophorese aufgelöst. Real-time-PCR (RT-PCR) wurde durchgeführt, um zu quantifizieren, die Prozent der Löschung Verwendung von Primern, die über der Kreuzung oder innerhalb der Löschung region. Die vergleichende Cq-Methode wurde verwendet, um den Expressionsgrad der Zielregion relativ zu einer Referenzregion (ACTB Locus) zu berechnen., Prozent der Deletion in der Zielregion wurde weiter durch das Verhältnis der Zielzellen relativ zu Kontrollzellen berechnet. Alle primer-Sequenzen aufgeführt sind in der Ergänzenden Tabelle S2.
Zielsequenzierung
Zellen wurden zwei Tage nach der Transfektion geerntet und die genomische DNA wurde mit QuickExtract DNA Extraction Solution (Epicentre Biotechnologies) extrahiert. PCR wurde durchgeführt, um die Zielregion mit genomischer DNA aus den Zellen zu amplifizieren, und Amplicons wurden von MiSeq Personal Sequencer (Illumina, San Diego, CA) tief sequenziert.,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Die Spendersequenzen werden im ergänzenden Material dargestellt. Die beiden 5‘ – meisten Verbindungen sind Phosphorothioat (durch Sternchen gekennzeichnet). Zellen in 6-well-Platten wurden transient transfiziert mit 2,0 µg Cas9-plasmid, 0.8 µg gRNA T1 plasmid, 0.8 µg gRNA T2 plasmid, und 0,4 µg linearer Geber Verwendung von Lipofectamine 2000 (Invitrogen). Nach 48 h nach der Transfektion wurden die Zellen 24 h lang mit 1 ng/ml TNF-α behandelt und dann GFP-positive Zellen sortiert.
Luciferase-Assay
Für den Luciferase-Assay wurden HEK 293T-Zellen in 96-Well-Platten mit einer Dichte von 5000 Zellen pro Well ausgesät., Nach 24 h wurden die Zellen transient transfiziert wurden mit 5 ng pRL-TK-Renilla-luciferase-reporter und 100 ng luciferase-reporter mit cytomegalovirus (CMV), SV40 (Simian virus 40), oder basic-Veranstalter. Nach 48 h wurde die Luciferase-Aktivität mit dem dualen Luciferase Reporter Assay System (Promega) gemessen.
Western blot
Proteine wurden durch Natriumdodecylsulfat—PAGE (SDS-PAGE) getrennt und auf Nitrozellulosemembranen übertragen. Die Membranen wurden mit 5% fettfreier Milch blockiert und mit GFP-Antikörper inkubiert (CST, #2555S, Danvers, MA)., Der Antigen-Antikörper-Komplex wurde mit verbesserten Chemilumineszenzreagenzien nachgewiesen.
Ergebnisse und Diskussion
Wir haben das bakterielle CRISPR / Cas9-System vom Typ II angepasst, um genomische DNA in menschlichen Zellen zu mutagenisieren. Die humane Codon-optimierte Version des S. pyogenes Cas9-Proteins mit einem C-Terminus SV40-Kernlokalisationssignal wurde unter Verwendung eines zuvor beschriebenen Systems exprimiert (6). Um die Cas9-Spaltung auf die gewünschte Sequenz zu lenken, exprimierten wir crRNA-tracrRNA-Fusionstranskripte, im Folgenden als Leitfaden-RNAs (gRNAs) bezeichnet, von einem modifizierten humanen H1-Polymerase-III-Promotor., Das 3 ‚ – Ende des H1-Promotors wurde modifiziert, um die Transkription von gRNAs zu ermöglichen, die mit einem Nukleotid beginnen. Eingeschränkt nur durch die Anforderung, dass dem 20 bp crRNA-Ziel die PAM-Sequenz NGG folgt (wobei N ein Nukleotid ist), kann dieser Ansatz prinzipiell verwendet werden, um auf jeden genomischen Ort mit der Form N20NGG abzuzielen. Um das Klonen des gRNA-Expressionsvektors zu erleichtern, verwendeten wir ein Typ-IIS-Restriktionsenzym, BsaI. Dies erforderte die Synthese eines 24 bp-Oligonukleotids, das einen Komplementaritätsbereich zur Zielstelle auf der DNA enthielt., Das einfache und effiziente Protokoll zum Klonen des gRNA-Expressionsvektors (Abbildung 1A) ist im Ergänzungsmaterial ausführlich beschrieben.
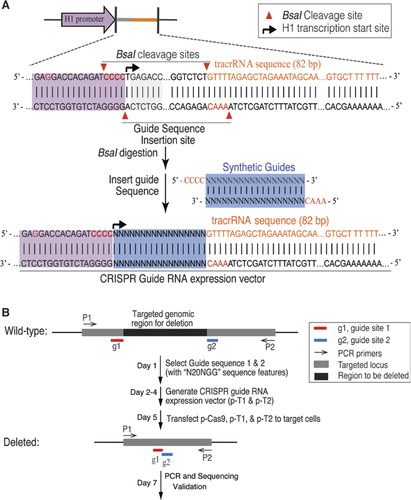
(A) Design des Guide RNA (gRNA) Expressionsvektors. Der Vektor wurde entwickelt, um gRNA-Transkripte mit einer synthetischen gRNA zu erzeugen, die mit der transaktivierenden RNA/CRISPR-RNA (tracrRNA) verschmolzen ist., Der H1-Promotor wurde modifiziert, um die interne BsaI-Stelle des Typ-IIS-Restriktionsenzyms zu eliminieren, indem GAGACC im H1-Promotor in GAGGACC geändert wurde. Eine BsaI-Site wurde eingeführt, um Klonstellen für gRNA – und tracrRNA-Fusionen durch Einfügen synthetischer Oligonukleotid-Duplexe mit kompatiblen Überhängen zu erstellen. Das 3 ‚ – Ende des H1-Promotors wurde modifiziert, um CCACAGATCCCC zu sein, um die Transkription von gRNAs mit einem Nukleotid am 5‘ – Ende zu erleichtern. (B) Die Schritte der gezielten Gen-Deletion mit CRISPR/Cas9.,
Zum Löschen eines großen Segments genomischer DNA verwendeten wir ein gRNAs-Paar gegen den Zielort (Abbildung 1B). Zwei Zielstellen mit dem Muster N20NGG wurden an der Grenze der Zielregion ausgewählt. Die Wirkungsgrade der gezielten Deletion, die von verschiedenen Kombinationen von gRNA-Paaren geleitet wurden, wurden durch PCR-Analysen unter Verwendung von Primern bestimmt, die die Zielregionen flankieren. Wildtyp – und abgeschnittene Genomfragmente wurden durch Gelelektrophorese aufgelöst. Um eine PCR-Amplifikations-Verzerrung zu vermeiden, wurde die prozentuale Deletion durch RT-PCR mit einem Primerpaar quantifiziert., Die Primer wurden über den Löschknotenpunkt (einen Primer außerhalb des Löschbereichs, den anderen Primer innerhalb des Löschbereichs) oder innerhalb der Löschbereiche (beide Primer befinden sich innerhalb des Löschbereichs) ausgelegt. Somit wird nur ein einziges Band mit dem Primerpaar sowohl für die Zielzellen als auch für Kontrollzellen amplifiziert. Wir berechneten die prozentuale Deletion durch Vergleich der relativen Menge an PCR-Produkten (Zielzellen versus Kontrollzellen), die durch dasselbe Primerpaar amplifiziert wurden. Gezielte Löschungen wurden durch Sequenzierung weiter verifiziert.,
Um zu beurteilen, wie sich gRNA-Paare auf die nachfolgende Reparatur und Erzeugung von Deletionen auswirken könnten, entwarfen wir zunächst gRNA-Sätze, die auf den genomischen Ort des menschlichen CDC42 abzielen und durch Entfernungen von etwa 200 bis 10.000 bp getrennt sind (Abbildung 2A und ergänzende Tabelle S1). Wir bewerteten dann die Fähigkeit jedes gRNA-Paares, Deletionen in menschlichen HEK 293T-Zellen in Gegenwart von Cas9 zu erzeugen. Robuste Wirkungsgrade von Löschungen auf NHEJ-Basis (bis zu 68%) wurden durch qPCR bestätigt (Abbildung 2B–2D)., Selbst für die Deletion einer 10 kb genomischen Region erhielten wir Targeting-Raten von 16% bis 28%, abhängig vom gRNA-Paar. Dieser RNA-vermittelte Bearbeitungsprozess war schnell, wobei die erste nachweisbare Deletion ungefähr 12 h nach der Transfektion auftrat (ergänzende Abbildung S1). Das System war in einer Vielzahl von Zelltypen wirksam, einschließlich: PC3 -, SK-Hep1-und HeLa-Zellen (ergänzende Abbildung S2).

(A) Schematische Darstellung der Standorte von Leitungs-RNAs (gRNAs), die auf den CDC42-Locus abzielen. (BD) Die Effizienz der gezielten Deletion mit CRISPR/Cas9 wurde durch PCR in HEK 293T-Zellen bestimmt. Primer außerhalb der erwarteten Löschregionen wurden verwendet. Der Prozentsatz der Löschung wurde durch RT-PCR unter Verwendung von Primern über die Kreuzung oder innerhalb der Löschregion quantifiziert. (E–F) Die Effizienz und Präzision der gezielten Deletion in CDC42 wurde durch (E) Sanger-Sequenzierung und (F) Hochdurchsatz-Sequenzierungsanalysen bestätigt. Das PCR-Produkt, das nur das Deletionsamplicon enthielt, wurde zur Sequenzierung angereichert.,
Die Löschungen wurden durch Sequenzierung von PCR-Produkten an den erwarteten Spaltstellen weiter bestätigt. Die Sanger-Sequenzierung zeigte, dass Deletionsknoten aus der genauen Ligation der von Cas9 erzeugten stumpfen DSBs resultierten; Jeder DSB trat genau 3 bp vor der PAM-Sequenz auf (Abbildung 2E und ergänzende Abbildung S3). Wir verwendeten auch eine tiefe Sequenzierung von Deletionsamplikonen, um die Genauigkeit der Deletionseffizienz zu beurteilen; In etwa 80% der Lesevorgänge wurden die gezielten DSBs perfekt repariert (Abbildung 2F).,
Wir haben diese Ergebnisse rekapituliert, indem wir gRNA-Paare getestet haben, die Fragmente aus einem genomischen Ort löschen, der das microRNA miR-21-Gen in HEK 293T-Zellen enthält. Zwei gRNAs wurden entwickelt, um auf die Grenzen der miR-21-Haarnadel abzuzielen (ergänzende Abbildung S4). Die Deletionseffizienz betrug 38% nach Transfektion mit den beiden gRNAs und Cas9, gemessen unter Verwendung eines PCR-Assays (ergänzende Abbildung S4B). Eine tiefe Sequenzierung bestätigte, dass die Löschung genau wie erwartet erfolgte (ergänzende Abbildung S4C).,
Um zu untersuchen, ob das CRISPR / Cas9-vermittelte Gen-Deletionssystem durch den Transkriptionszustand gezielter Gene beeinflusst wird, wurde das für den Chemokin-Liganden 2 (CCL2) codierende Gen gezielt. CCL2 ist ein kleines Zytokin, das zur CC-Chemokinfamilie gehört; Das CCL2-Gen ist ein Ziel der NF-kB-Signalisierung. Wir haben zufällig acht Zielstellen ausgewählt, die sich an den 5‘ und 3‘ Enden des CCL2-Genlocus befinden (Abbildung 3A). Wir haben eine robuste und effiziente Deletion verschiedener Regionen des Gens unter Verwendung von Cas9 und verschiedenen gRNAs-Paaren in HEK 293T-Zellen erreicht (Abbildung 3B-3D).,
(A) Schematische Darstellung der Positionen von Leitungs-RNAs (gRNAs), die auf den CCL2-Locus abzielen. (B–D) Die Effizienz der gezielten Löschung von CCL2 mit CRISPR/Cas9 in HEK 293T-Zellen. (E) Die CCL2-mRNA-Spiegel wurden durch Zugabe von Tumornekrosefaktor α (TNF-α) durch quantitative RT-PCR-Analysen in HEK 293T-Zellen bestimmt. Die Daten wurden mit dem Mittel ± sem in dreifacher Ausfertigung gezeigt., (F) Die Effizienz der gezielten Deletion von CCL2 mit CRISPR/Cas9 nach Behandlung mit TNF-α für 24 h in HEK 293T-Zellen. (G) Das Niveau der Luciferase-Aktivität von Cytomegalovirus (CMV) oder SV40 oder einem basischen Promotor in HEK 293T-Zellen. Die Daten wurden mit dem Mittel ± sem in dreifacher Ausfertigung gezeigt. (H) Effizienz der gezielten Deletion eines Luciferase-Gens, das durch CMV oder SV40 oder einen basischen Promotor in HEK 293T-Zellen gesteuert wird. (B-D,F, H) Der Prozentsatz der Löschung wurde durch RT-PCR unter Verwendung von Primern über die Kreuzung oder innerhalb der Löschregion quantifiziert.,
Die Expression des CCL2-Gens wurde durch TNF-α (bis zu ∼300-facher Anstieg) in HEK 293T-Zellen (Abbildung 3E) dramatisch induziert und lieferte ein gutes Modell, um zu untersuchen, ob die CRISPR/ Cas9-vermittelte gezielte Genombearbeitung durch Transkriptionsaktivität beeinflusst wird. Interessanterweise wurde die Effizienz der gezielten Deletion des CCL2-Genlocus durch die Behandlung von Zellen mit TNF-α nicht beeinflusst(Abbildung 3F), was darauf hindeutet, dass die Transkription die CRISPR/Cas9-vermittelte Deletion nicht veränderte., Um dieses Ergebnis weiter zu bestätigen, zielten wir auf ein exogenes Reportergen ab, das von verschiedenen Promotoren mit unterschiedlichen Stärken angetrieben wurde, wobei die verschiedenen Transkriptionsaktivitäten unter Verwendung eines Luciferase-Assays bewertet werden konnten (Abbildung 3G). PCR-Assays zeigten eine ähnliche Effizienz bei gezielten Deletionen in HEK 293T-Zellen nach Co-Transfektion des Reportergens zusammen mit Cas9-und gRNA-Paaren (Abbildung 3H). Dieses Ergebnis zeigt an, dass eine NHEJ-vermittelte Reparatur trotz des Auftretens unterschiedlicher Transkriptionsaktivität auftreten kann.,
DSBs können HDR stimulieren, um einen hochpräzisen Ersatz der beschädigten Region durch einen homologen Spender zu ermöglichen. Um einen gezielten genomischen DNA-Ersatz zu erhalten, haben wir ein Paar gRNAs, Cas9 und einen linearen Spender mit Homologie in die Zielregion in Zellen eingeführt (Abbildung 4A). Der lineare Donor wurde durch PCR-Amplifikation mit Primern mit einer homologen Sequenz von 50 bp erhalten. Derselbe Spender wurde erfolgreich mit einem ZFN-basierten HDR-Reparatursystem (18) eingesetzt., Um die Machbarkeit eines CRISPR / Cas9-vermittelten Ersetzens durch HDR zu testen, zielten wir auf den CCL2-Locus mit einem Paar gRNAs (#39 und #1854 in Abbildung 3) und einem Spender mit der Kodierungssequenz Enhanced Green Fluorescent Protein (EGFP) und der SV40 Poly(A) – Stelle (Abbildung 4A; Sequenzen und Positionen sind im Ergänzungsmaterial dargestellt). Unter Verwendung dieses Systems waren etwa 0,5% der Zielzellen EGFP-positiv, während nur 0,023% EGFP-positiv in Scheintransfektionszellen waren (nur mit Spender transfektiert), was den Kontrollzellen ähnlich war (0,021%, ohne Transfektion)., Die EGFP-positiven Zellen wurden dann durch Durchflusszytometrie sortiert. Die ortsspezifische Integration wurde durch PCR unter Verwendung von zwei Primerpaaren bestätigt, die sowohl die homologen Arme als auch die gesamte ersetzte Region flankierten. Wie in Abbildung 4B gezeigt, beobachteten wir den erwarteten ersetzten Bereich, der die EGFP-Sequenz in voller Länge und die homologen Arme enthielt (Sangersequenzierungsergebnis im Ergänzungsmaterial). Das endogene Wildtyp-Allel wurde ebenfalls nachgewiesen (Abbildung 4B), was darauf hinweist, dass nicht alle Allele gezielt sind., Darüber hinaus wählten wir einzelne Klone aus den EGFP-positiven Zellen aus und stellten fest, dass alle Klone (6 von 6 untersuchten) die erwartete Integration hatten (Abbildung 4C), aber das endogene Wildtyp-Allel wurde auch in drei der Klone nachgewiesen (Abbildung 4C), was darauf hindeutet, dass nur ein Allel in diesen Klonen gezielt war. Die Expression von EGFP-Protein in Zielzellen (EGFP-positive sortierte Zellen) wurde auf TNF-α-Behandlung up-reguliert, wie durch Western Blot und Fluoreszenz aktivierte Zellsortierung (FACS) ausgewertet (Abbildung 4, D und E)., Diese Ergebnisse zeigten, dass das CRISPR/Cas9-System verwendet werden kann, um Gen – /Domänenersatz mit hoher Effizienz und Genauigkeit zu erstellen.

(A) Schematische Diagramme, die das Verfahren des gezielten Genersatzes unter Verwendung von CRISPR/Cas9 in menschlichen Zellen darstellen., Um die Wirksamkeit eines gezielten Genersatzes zu testen, wurden Guide RNAs (gRNAs) entwickelt, um die angegebene Region (markiert durch die Stellen 1 und 2) des CCL2-Gens zu löschen und die gelöschte Region durch den EGFP-polyA-Kassettenspender durch Arme mit kurzen homologischen Regionen zu ersetzen. Die Zielstellen von Site 1 und Site 2 sind #39 und #1854 innerhalb des CCL2-Gens in Abbildung 3. Die homologen Sequenzen (50 bp) sind nur stromaufwärts und stromabwärts der Löschstellen. Die Positionen und Sequenzen im Detail sind im ergänzenden Material dargestellt., (B) Die Effizienz des gezielten Genersatzes mit CRISPR/Cas9 wurde durch PCR in HEK 293T-Zellen bestimmt. Primer, die die Übergänge zwischen CCL2 und EGFP überspannten, wurden für die PCR-Verstärkung verwendet. (C) PCR-Assay für den gezielten Genersatz der einzelnen Klone. (D,E) Die Expression von EGFP-Protein bei TNF-α-Addition in HEK 293T-Zellen wurde durch (D) Western Blot und (E) FACS bestimmt.
Hier beschreiben wir einen einfachen und effizienten Ansatz zur Gen-Deletion mit dem CRISPR/Cas9-System., Wir haben gezeigt, dass die Einführung dieses Systems in menschliche HEK 293T-Zellen und andere menschliche Zelltypen Deletionen von Fragmenten bis zu 10 kb mit Wirkungsgraden zwischen 11% und 68% induzierte, abhängig von der Zielsequenz. Die Fähigkeit, genomische Segmente effizient und präzise zu löschen, erleichtert die Untersuchung funktioneller genomischer Elemente in menschlichen Zellen. Dieser Ansatz kann möglicherweise verwendet werden, um auf alle genomischen Loci abzuzielen.
Es gab Bedenken hinsichtlich der Spezifität des CRISPR / Cas9-Systems (19-21)., Um unerwünschte Phänotypen aufgrund von Off-Target-Mutationen auszuschließen, empfehlen wir, für jede Zielregion mindestens zwei verschiedene gRNAs-Paare zu verwenden. In unserer Studie war der Bedarf an mehreren gRNA-Paaren angesichts der Einfachheit und hohen Effizienz dieses Systems keine wesentliche Einschränkung. Es ist bemerkenswert, dass verschiedene gRNAs-Paare, die auf dieselbe Region abzielen, mit hoher Effizienz arbeiteten (Abbildungen 2 und 4). Ein weiterer Ansatz zur Vermeidung unerwünschter Mutationen ist die Verwendung der Double Nickase-Methode (22, 23)., Wir haben auch die Double Nickase-Methode erfolgreich angewendet, um eine Deletion genomischer DNA zu erzeugen, aber die Effizienz war erheblich geringer.
Es ist bekannt, dass die Reparatur von DNA-DSBs weitgehend durch fehleranfälliges NHEJ vermittelt wird, bei dem die beiden Enden auf eine Weise verarbeitet und ligiert werden, die häufig von Nukleotideinfügungen und-löschungen begleitet wird. Eine solche fehleranfällige Endverbindung wurde bei der Reparatur von DSBs beobachtet, die von ZFNs oder TALENs erstellt wurden. Im Gegensatz dazu war die Reparatur von DSBs, die von Cas9 und zwei gRNAs erzeugt wurden, sehr präzise., Unsere Ergebnisse legen nahe, dass die Pausen ohne Endverarbeitung direkt ligiert werden, was einen zuvor nicht geschätzten Vorteil des NHEJ-Pfades offenbart. Der Mechanismus, der zu den genauen Ligationen führt, bleibt zu bestimmen. Eine Möglichkeit besteht darin, dass das gezielte Löschen mit Cas9 und zwei gRNAs zu einer Kreuzung führt, die von keinem der ursprünglichen gRNAs erkannt wird. Wir analysierten auch die Effizienz bei der Erzeugung von Indel-Mutationen für das einzelne gRNA – und gRNA-Paar (#39 und #224 gRNA in Abbildung 3A) durch Sanger-Sequenzierung von PCR-Amplikons (TA-Klonen)., Von Interesse beobachteten wir, dass die Effizienz der Erzeugung von Indel-Mutationen für einzelne gRNA ziemlich gering war (9,5%, 2 von 21 Klonen für #39 gRNA; 5%, 1 von 20 Klonen für #224 gRNA). Das gRNA-Paar erzeugte jedoch eine hohe Effizienz von Indel-Mutationen (50%, 10 von 20 Klonen für #39 und #224), was dem Assay mit qPCR ähnlich war (52%, Abbildung 3B). Wir schlagen vor, dass ein einzelner gRNA oft zu einem stumpfen Ende der Spaltstelle führt, das von NHEJ genau repariert wird. Somit ist die Effizienz der Erzeugung von Mutationen bei Verwendung einer einzelnen gRNA viel geringer als bei einem gRNAs-Paar.,
Anerkennungen
Diese Arbeit wurde durch Zuschüsse des National Institute of Health (Nr. DP1CA174421) und der W. M. Keck-Stiftung, C.-Z. C, und die National Natural Science Foundation of China (No. 81101481) und Shanghai Medizinisches Talent-Training-Programm (Nr. XYQ2011048) an S. L. H. Dieses Papier unterliegt der NIH Public Access Policy.
Konkurrierende Interessen
Die Autoren erklären keine konkurrierenden Interessen.
Ergänzende Daten
Um die ergänzenden Daten zu diesem Papier anzuzeigen, besuchen Sie bitte die Website der Zeitschrift unter: www.,future-science.com/doi/suppl/10.2144/000114196