Eliminación precisa de genes y reemplazo utilizando el sistema CRISPR / Cas9 en células humanas
Aquí mostramos que dos ARN guía junto con Cas9 generan eficientemente deleciones de ADN de hasta 10 kb en células humanas en un proceso donde la reparación de la deleción se logra en gran medida mediante la Unión final precisa. Además, proporcionamos datos que muestran que el sistema CRISPR / Cas9 puede reemplazar grandes fragmentos genómicos en presencia de un donante de reparación homólogo lineal.,
los loci bacterianos agrupados regularmente interespaciados de repeticiones palindrómicas cortas/asociadas a CRISPR (CRISPR / Cas) codifican sistemas inmunitarios guiados por ARN que protegen a las células contra virus invasores y plásmidos (1, 2). En Streptococcus pyogenes, los sistemas CRISPR/Cas tipo II utilizan una endonucleasa guiada por ARN (RGEN), Cas9, para catalizar la escisión específica del sitio de las secuencias de ADN Diana., La orientación de Cas9 a sitios genómicos específicos está mediada por una secuencia guía de 20 nucleótidos dentro de un ARN CRISPR asociado (arncr) y requiere un ARNr transactivador (arnrtr) que recluta el arnrr en el complejo Cas9 (3). El reconocimiento de los sitios de escisión está determinado por el emparejamiento de bases de ADN-ARNr y un motivo adyacente a protospacer (PAM), una secuencia de tres nucleótidos (NGG) yuxtapuesta a la región complementaria del ADN (4)., Es notable que un ARN guía único (gRNA) que imita el complejo tracrRNA-crRNA puede reclutar Cas9 para sitios genómicos específicos y generar roturas de doble cadena (DSBs) en el ADN (5). Los sistemas CRISPR/Cas9 se han adaptado para la edición genómica específica del sitio en diversos tipos de células y organismos (6-12).
La edición del genoma con CRISPR / Cas9 se inicia con la introducción de un DSB en un locus genómico objetivo utilizando el RGEN programado por ARN. Esto es seguido por la reparación del DSB a través de la reparación dirigida por homología (HDR) o la Unión final no homóloga (NHEJ)., En presencia de un donante de reparación homólogo, el sistema CRISPR/Cas9 puede utilizarse para generar modificaciones e inserciones precisas y definidas en un locus objetivo a través del proceso HDR. En ausencia de un donante de reparación homólogo, los DSB únicos generados por CRISPR/Cas9 se reparan a través del nhej propenso a errores, lo que resulta en mutaciones de inserción o deleción (indel). Las mutaciones Indel en los exones codificantes pueden introducir codones de parada prematuros o mutaciones de cambio de Marco, inactivando así las proteínas correspondientes., Las mutaciones de Indel generadas por la reparación de un único DSB pueden no ser útiles en experimentos destinados a caracterizar los dominios funcionales de los genes codificadores de proteínas o para la inactivación de elementos genómicos tales como secuencias reguladoras intergenicas o intrónicas o genes de ARN no codificantes. Las eliminaciones de fragmentos de ADN en los loci objetivo proporcionarían una vía para estudiar estos elementos funcionales. Con este fin, se han introducido múltiples DSBs para generar deleciones en Drosophila (12, 13), pez cebra (14) y células humanas (8), aunque con baja eficiencia., También se han logrado deleciones genómicas específicas de ADN utilizando nucleasa de dedo de zinc (ZFN) o nucleasa efectora tipo activador de transcripción (TALEN) en células humanas (15-17). Sin embargo, las eficiencias de estos enfoques son generalmente bajas. Además, ZFNs y TALENs siguen siendo algo difíciles y costosos de diseñar, desarrollar y probar empíricamente en el contexto celular.
aquí, examinamos la generación de deleciones en células humanas catalizadas por el sistema CRISPR / Cas9. Mostramos que 2 gRNAs junto con Cas9 pueden crear eficientemente deleciones de ADN de hasta 10 kb., De interés, encontramos que la reparación de este proceso de eliminación se logra en gran medida mediante la Unión final precisa. Además, la supresión dirigida con CRISPR/Cas9 parece ser independiente del estado transcripcional del locus objetivo. Finalmente, mostramos que el sistema CRISPR / Cas9 puede ser utilizado para reemplazar grandes fragmentos genómicos en presencia de un donante de reparación homólogo lineal.
Material y métodos
construcción del plásmido
el promotor básico de H1 se amplificó a partir del plvthm plásmido (Addgene, #12247, Cambridge, MA)., Se sintetizaron oligonucleótidos que contenían el promotor H1 modificado y la columna vertebral de las secuencias de gRNA deseadas con dos sitios de BsaI (centro PAN, Universidad de Stanford). Los productos de longitud completa resultantes fueron amplificados por PCR y clonados en el vector pUC19. El gen ampicilina (amp) y el promotor H1 en el vector pUC19 contienen sitios enzimáticos de restricción BsaI; estos fueron mutados (el gen amp fue cambiado de G1601C, que no altera la secuencia de aminoácidos; el promotor H1 fue cambiado de GAGACC a GAGGACC) para eliminar los sitios BsaI., El protocolo para la clonación gRNA se presenta en el material complementario. Todas las secuencias de los sitios de destino se presentan en el cuadro suplementario S1.
cultivo celular
se cultivaron células HEK 293T, SK-Hep1 y hela en medio de Dulbecco»s modified Eagle»s (DMEM) suplementado con suero fetal bovino (FBS) al 10% (Hyclone, Logan, UT) y penicilina/estreptomicina (pen/strep) (Invitrogen, Carlsbad, CA). Se cultivaron células PC3 en medio RPMI-1640 suplementado con 10% de FBS y pen/strep., Para la estimulación del factor de necrosis tumoral α (TNF-α), se trataron 293 células T con concentraciones indicadas de TNF-α(r&D Systems, Minneapolis, MN). Las células se mantuvieron a 37 ° C y 5% de CO2 en una incubadora humidificada.
deleción de ADN dirigida
las células HEK 293T se sembraron en placas de 12 pocillos a una densidad de 100.000 células por pocillo. Después de 24 h, las células se transfectaron transitoriamente con plásmidos de 1 µg Cas9 (Addgene, #41815), 0.5 µg gRNA T1 y 0.5 µg grna T2 usando Lipofectamina 2000 (Invitrogen) según los protocolos del fabricante., El ADN genómico se extrajo 48 h después de la transfección utilizando la solución de extracción de ADN QuickExtract (Epicentre Biotechnologies, Madison, WI). La PCR común se llevó a cabo para amplificar la región objetivo utilizando cebadores que flanqueaban las regiones objetivo. Los fragmentos genómicos truncados y de tipo salvaje se resolvieron mediante electroforesis en gel. La PCR en tiempo Real (RT-PCR) se realizó para cuantificar el porcentaje de eliminación utilizando cebadores a través de la Unión o dentro de la región de eliminación. Se utilizó el método comparativo Cq para calcular el nivel de expresión de la región objetivo en relación con una región de referencia (ACTB locus)., El porcentaje de deleción en la región objetivo se calculó además por la proporción de células objetivo en relación con las células de control. Todas las secuencias de imprimación se enumeran en la tabla suplementaria S2.
secuenciación objetivo
Las células se recolectaron dos días después de la transfección, y el ADN genómico se extrajo utilizando la solución de extracción de ADN QuickExtract (Epicentre Biotechnologies). La PCR se llevó a cabo para amplificar la región objetivo con ADN genómico derivado de las células, y los amplicones fueron secuenciados en profundidad por MiSeq personal Sequencer (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Las secuencias del donante se presentan en el material suplementario. Los dos enlaces 5 ‘ – mayoría son fosforotioato (indicado por asteriscos). Las células en placas de 6 pocillos fueron transfectadas transitoriamente con 2,0 µg de plásmido Cas9, 0,8 µg de plásmido gRNA T1, 0,8 µg de plásmido gRNA T2 y 0,4 µg de donante lineal usando Lipofectamina 2000 (Invitrogen). A las 48 h después de la transfección, las células se trataron con 1 ng/mL de TNF-α durante 24 h, y luego se clasificaron las células GFP positivas.
ensayo de luciferasa
para el ensayo de luciferasa, se sembraron células HEK 293T en placas de 96 pocillos a una densidad de 5000 células por pocillo., Después de 24 h, las células fueron transfectadas transitoriamente con 5 ng de PRL-TK Renilla luciferase reporter y 100 ng de luciferase reporter con citomegalovirus (CMV), SV40 (Virus simio 40), o promotor básico. Después de 48 h, la actividad de la luciferasa se midió con el Dual luciferase reporter assay system (Promega).
Western blot
Las proteínas fueron separadas por dodecil sulfato de sodio—PAGE (sds-PAGE) y transferidas a membranas de nitrocelulosa. Las membranas fueron bloqueadas con leche descremada al 5% e incubadas con anticuerpo GFP (CST, #2555S, Danvers, MA)., El complejo antígeno-anticuerpo se detectó con reactivos quimioluminiscentes mejorados.
resultados y discusión
adaptamos el sistema bacteriano tipo II CRISPR / Cas9 para mutagenizar ADN genómico en células humanas. La versión humana optimizada para codones de la proteína S. pyogenes Cas9 con una señal de localización nuclear C-terminus SV40 se expresó utilizando un sistema descrito anteriormente (6). Para dirigir la escisión Cas9 a la secuencia deseada, expresamos las transcripciones de fusión crRNA-tracrna, en lo sucesivo denominadas ARN guía (gRNAs), de un promotor humano modificado de la polimerasa III H1., El extremo 3′ del promotor H1 fue modificado para permitir la transcripción de gRNAs que comienzan con cualquier nucleótido. Limitado solo por el requisito de que el objetivo de 20 bp crRNA sea seguido por la secuencia PAM, NGG (donde N es cualquier nucleótido), este enfoque puede en principio ser utilizado para apuntar a cualquier ubicación genómica que tenga la forma N20NGG. Para facilitar la clonación del vector de expresión gRNA, utilizamos una enzima de restricción de tipo IIs, BsaI. Esto requirió la síntesis de un oligonucleótido de 24 PB que contenía una región de complementariedad con el Sitio objetivo en el ADN., El Protocolo simple y eficiente para la clonación del vector de expresión gRNA (figura 1a) se describe en detalle en el material suplementario.
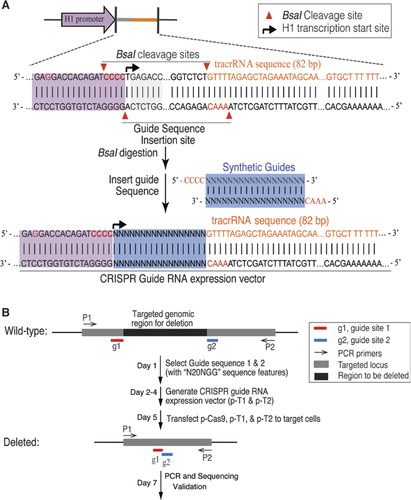
(A) Diseño del vector de expresión del ARN guía (gRNA). El vector fue diseñado para producir transcripciones de gRNA con un grna sintético fusionado al ARN transactivador / ARN CRISPR (tracrRNA)., El promotor H1 fue modificado para eliminar el sitio interno de la enzima de restricción IIS bsai cambiando GAGACC en el promotor H1 A GAGGACC. Se introdujo un sitio de BsaI para crear sitios de clonación para fusiones de gRNA y tracrRNA mediante la inserción de duplexes de oligonucleótidos sintéticos con voladizos compatibles. El extremo 3′ del promotor H1 fue modificado para ser CCACAGATCCCC para facilitar la transcripción de gRNAs con cualquier nucleótido en el extremo 5′. (B) los pasos de la deleción génica dirigida con CRISPR/Cas9.,
para la deleción de un segmento grande de ADN genómico, se utilizó un par de gRNAs contra el locus objetivo (figura 1B). Se seleccionaron dos sitios objetivo con el patrón N20NGG en el límite de la región objetivo. Las eficiencias de la deleción dirigida guiada por varias combinaciones de pares de gRNA se determinaron mediante análisis de PCR utilizando cebadores que flanquean las regiones objetivo. Los fragmentos genómicos truncados y de tipo salvaje se resolvieron mediante electroforesis en gel. Para evitar el sesgo de amplificación de la PCR, se cuantificó el porcentaje de deleción mediante RT-PCR utilizando un par de cebadores., Los cebadores se diseñaron a través de la Unión de eliminación (un cebador fuera de la región de eliminación, el otro cebador dentro de la región de eliminación) o dentro de las regiones de eliminación (ambos cebadores se ubican dentro de la región de eliminación). Por lo tanto, solo una sola banda se amplifica con el par de imprimación tanto para las células objetivo como para las células de control. Calculamos el porcentaje de eliminación comparando la cantidad relativa de productos de PCR (células objetivo versus células control) amplificados por el mismo par de imprimación. Las supresiones selectivas se verificaron mediante secuenciación.,
para evaluar cómo los pares de gRNA podrían afectar la reparación posterior y la generación de eliminaciones, primero diseñamos conjuntos de grna dirigidos contra el locus genómico humano CDC42 y separados por distancias que van desde aproximadamente 200 a 10.000 PB (figura 2a y tabla suplementaria S1). Luego evaluamos la capacidad de cada par de gRNA para generar deleciones en células HEK 293t humanas en presencia de Cas9. Las eficiencias robustas de las eliminaciones basadas en NHEJ (hasta el 68%) fueron confirmadas por el qPCR (figura 2B–2D)., Incluso para la eliminación de una región genómica de 10 kb, obtuvimos tasas de objetivo de 16% a 28%, dependiendo del par de gRNA. Este proceso de edición mediado por ARN fue rápido, con la primera deleción detectable apareciendo aproximadamente 12 h después de la transfección (figura suplementaria S1). El sistema fue eficaz en una variedad de tipos de células, incluyendo: PC3, SK-Hep1, y células HeLa (figura suplementaria S2).

(A) diagrama esquemático que representa la ubicación de los RNAs guía (gRNAs) dirigidos al locus CDC42. (B–D) la eficiencia de la deleción dirigida con CRISPR/Cas9 se determinó mediante PCR en células HEK 293t. Se utilizaron cebadores fuera de las regiones de eliminación previstas. El porcentaje de deleción se cuantificó mediante RT-PCR utilizando cebadores a través de la Unión o dentro de la región de deleción. (E-F) la eficiencia y precisión de la eliminación selectiva en CDC42 se confirmó mediante (e) secuenciación Sanger y (F) análisis de secuenciación de alto rendimiento. El producto de PCR que contenía solo el amplicón de deleción se enriqueció para la secuenciación.,
las eliminaciones se confirmaron mediante la secuenciación de productos de PCR que abarcaban los sitios de escisión esperados. La secuenciación de Sanger mostró uniones de deleción resultantes de la ligadura precisa de los DSBs de extremo romo creados por Cas9; cada DSB ocurrió exactamente 3 pb aguas arriba de la secuencia PAM (figura 2E y figura suplementaria S3). También utilizamos la secuenciación profunda de los amplicones de eliminación para evaluar la precisión de la eficiencia de la eliminación; en aproximadamente el 80% de las lecturas, los DSB objetivo se repararon perfectamente (figura 2F).,
recapitulamos estos hallazgos probando pares de gRNA diseñados para eliminar fragmentos de un locus genómico que contiene el gen microRNA miR-21 en células HEK 293t. Se diseñaron dos grna para apuntar a los límites de la horquilla mir-21 (figura suplementaria S4). La eficiencia de deleción fue del 38% después de la transfección con los dos ARN y Cas9, medida mediante un ensayo de PCR (figura suplementaria S4B). La secuenciación profunda confirmó que la eliminación ocurrió exactamente como se esperaba (figura suplementaria S4C).,
para investigar si el sistema de deleción génica mediada por CRISPR / Cas9 está influenciado por el estado transcripcional de los genes dirigidos, el gen que codifica la quimiocina (motivo C-C) ligando 2 (CCL2) fue dirigido. CCL2 es una pequeña citocina que pertenece a la familia de las quimioquinas CC; el gen CCL2 es un blanco de la señalización NF-kB. Se seleccionaron aleatoriamente ocho sitios objetivo ubicados en los extremos 5′ y 3 ‘ del locus del gen CCL2 (figura 3a). Se logró una deleción robusta y eficiente de diferentes regiones del gen utilizando Cas9 y diferentes pares de gRNAs en células HEK 293T (figura 3B–3D).,

(A) diagrama esquemático que representa la ubicación de los ARN guía (grna) dirigidos al locus CCL2. (B–D) la eficiencia de la deleción dirigida de CCL2 con CRISPR/Cas9 en células HEK 293T. E) los niveles de ARNm de CCL2 se determinaron mediante la adición del factor de necrosis tumoral α (TNF-α) mediante análisis cuantitativos de RT-PCR en células HEK 293T. Los datos se mostraron con los promedios ± sem por triplicado., F) la eficacia de la deleción dirigida de CCL2 con CRISPR/Cas9 después del tratamiento con TNF-α durante 24 h en células HEK 293T. G) el nivel de actividad de la luciferasa del citomegalovirus (CMV), o SV40, o un promotor básico en las células HEK 293T. Los datos se mostraron con los promedios ± sem por triplicado. (H) eficiencia de la deleción dirigida de un gen de luciferasa controlado por CMV, o SV40, o un promotor básico en células HEK 293t. (B-D, F, H) el porcentaje de deleción se cuantificó mediante RT-PCR utilizando cebadores a través de la Unión o dentro de la región de deleción.,
la expresión del gen ccl2 fue dramáticamente inducida por el TNF-α (aumento de hasta 3 300 veces) en células HEK 293T (figura 3E), proporcionando un buen modelo para investigar si la edición del genoma dirigido mediada por CRISPR / Cas9 se ve afectada por la actividad transcripcional. De interés, la eficiencia de la deleción dirigida del locus del gen CCL2 no se vio afectada por el tratamiento de las células con TNF-α(figura 3F), lo que sugiere que la transcripción no alteró la deleción mediada por CRISPR/Cas9., Para confirmar aún más este resultado, nos dirigimos a un gen reportero exógeno impulsado por varios promotores con diferentes fortalezas, donde las diferentes actividades transcripcionales podrían evaluarse utilizando un ensayo de luciferasa (figura 3G). Los ensayos de PCR revelaron una eficiencia similar en las deleciones dirigidas en células HEK 293T después de la co-transfección del gen reportero junto con pares Cas9 y gRNA (figura 3H). Este resultado indica que la reparación mediada por NHEJ puede ocurrir a pesar de la ocurrencia de diversos grados de actividad transcripcional.,
el DSBs puede estimular el HDR para permitir el reemplazo altamente preciso de la región dañada con un donante homólogo. Para obtener el reemplazo de ADN genómico dirigido, se introdujo un par de gRNAs, Cas9 y un donante lineal con homología a la región objetivo en las células (figura 4a). El donante lineal se obtuvo por amplificación PCR con cebadores con una secuencia homóloga de 50 PB. Este mismo donante se insertó con éxito utilizando un sistema de reparación HDR basado en ZFN (18)., Para probar la viabilidad del reemplazo mediado por CRISPR/ Cas9 por HDR, apuntamos el locus CCL2 con un par de gRNAs (#39 y #1854 mostrados en la Figura 3) y un donante que lleva la secuencia codificante de proteína verde fluorescente mejorada (EGFP) y el sitio de poli(a) SV40 (figura 4A; las secuencias y posiciones se presentan en el Material suplementario). Usando este sistema, aproximadamente el 0,5% de las células dirigidas fueron EGFP-positivas, mientras que solo el 0,023% fueron EGFP-positivas en las células de transfección simuladas (solo transfectadas con el donante), que fue similar a las células de control (0,021%, sin transfección)., Las células EGFP positivas se clasificaron por citometría de flujo. La integración Site-specific fue confirmada por PCR usando dos pares de cebadores flanqueando tanto los brazos homólogos como toda la región reemplazada. Como se muestra en la figura 4B, observamos la región reemplazada esperada que contiene la secuencia EGFP de longitud completa y los brazos homólogos (el resultado de la secuenciación de Sanger se muestra en el Material suplementario). También se detectó el alelo endógeno de tipo silvestre (figura 4B), lo que indica que no todos los alelos están dirigidos., Además, seleccionamos clones individuales de las células EGFP positivas y encontramos que todos los clones (6 de 6 examinados) tenían la integración esperada (figura 4C), pero el alelo endógeno de tipo salvaje también se detectó en tres de los clones (figura 4C), lo que sugiere que solo un alelo fue objetivo en esos clones. La expresión de la proteína EGFP en células objetivo (células clasificadas EGFP-positivas) fue regulada al alza en el tratamiento con TNF-α evaluado por Western blot y clasificación celular activada por fluorescencia (FACS) (Figura 4, D y E)., Estos resultados demostraron que el sistema CRISPR / Cas9 se puede utilizar para crear reemplazos de genes/dominios con alta eficiencia y precisión.

(a) diagramas esquemáticos que representan el procedimiento de reemplazo génico dirigido utilizando CRISPR/Cas9 en células humanas., Para probar la eficacia del reemplazo génico dirigido, se diseñaron ARN guía (gRNAs) para eliminar la región indicada (marcada por los sitios 1 y 2) del gen CCL2 y para reemplazar la región eliminada con el donante de casete EGFP-polyA con brazos con regiones cortas de homología. Los sitios objetivo del Sitio 1 y el sitio 2 son #39 y #1854 dentro del gen CCL2 mostrado en la Figura 3. Las secuencias homólogas (50 PB) están justo aguas arriba y aguas abajo de los sitios de eliminación. Las posiciones y secuencias en detalle se presentan en el Material Complementario., B) La eficacia del reemplazo de genes específicos por CRISPR/Cas9 se determinó mediante PCR en células HEK 293T. Para la amplificación de PCR se utilizaron cebadores que abarcan las uniones entre CCL2 y EGFP. C) ensayo de PCR para el reemplazo de genes específicos de los clones individuales. (D,E) La expresión de la proteína EGFP tras la adición de TNF-α en las células HEK 293T se determinó mediante (D) Western blot y (E) FACS.
aquí describimos un enfoque simple y eficiente para la deleción génica utilizando el sistema CRISPR / Cas9., Demostramos que la introducción de este sistema en las células HEK 293t humanas, y otros tipos de células humanas, indujo supresiones de fragmentos de hasta 10 kb con eficiencias que oscilan entre el 11% y el 68%, dependiendo de la secuencia objetivo. La capacidad de eliminar de manera eficiente y precisa los segmentos genómicos facilitará el estudio de los elementos genómicos funcionales en las células humanas. Este enfoque puede ser potencialmente utilizado para apuntar a cualquier LOCI genómico.
ha habido preocupación con respecto a la especificidad del sistema CRISPR/Cas9 (19-21)., Para descartar fenotipos no deseados debido a mutaciones fuera del objetivo, sugerimos que se utilicen al menos dos pares diferentes de gRNAs para cada región objetivo. En nuestro estudio, la necesidad de múltiples pares de gRNA no fue una limitación importante, dada la simplicidad y alta eficiencia de este sistema. Es notable que diferentes pares de grna dirigidos a la misma región trabajaron con alta eficiencia (Figuras 2 y 4). Otro enfoque para evitar mutaciones no deseadas es el uso del método de doble nickase (22, 23)., También aplicamos con éxito el método de doble nickase para generar deleción de ADN genómico, pero la eficiencia fue considerablemente menor.
se sabe que la reparación de DNA DSBs está mediada en gran medida por nhej propenso a errores, en el que los dos extremos se procesan y ligan juntos de una manera que frecuentemente se acompaña de inserciones y deleciones de nucleótidos. Tal unión de extremos propensa a errores se observó en la reparación de DSBs creados por ZFNs o TALENs. Por el contrario, la reparación de los DSB generados por Cas9 y dos gRNAs fue muy precisa., Nuestros resultados sugieren que las rupturas se ligan directamente sin procesamiento final, revelando una ventaja previamente no apreciada de la vía NHEJ. El mecanismo que resulta en las ligaduras precisas queda por determinar. Una posibilidad es que la eliminación dirigida usando Cas9 y dos gRNAs resulta en una unión que no es reconocida por ninguno de los gRNAs originales. También analizamos la eficiencia en la generación de mutaciones indel para el gRNA individual y par de gRNA (#39 y # 224 gRNA en la figura 3a) mediante secuenciación Sanger de amplicones PCR (clonación TA)., De interés, observamos que la eficiencia de generación de mutaciones indel para un solo gRNA fue bastante baja (9.5%, 2 de 21 clones para el #39 gRNA; 5%, 1 de 20 clones para el #224 gRNA). Sin embargo, el par gRNA generó una alta eficiencia de mutaciones en indel (50%, 10 de 20 clones para #39 y #224), que fue similar al ensayo usando qPCR (52%, figura 3B). Proponemos que un solo gRNA a menudo resulta en un extremo romo del sitio de escisión, que será reparado con precisión por NHEJ. Por lo tanto, la eficiencia de generar mutación es mucho menor con el uso de un solo gRNA que un par de gRNAs.,
agradecimientos
Este trabajo fue apoyado por becas del Instituto Nacional de salud (No. DP1CA174421) y la Fundación W. M. Keck a C.-Z. C, y la Fundación Nacional de Ciencias Naturales de China (no. 81101481) y el programa de capacitación de talentos médicos de Shanghai (No. XYQ2011048) a S. L. H. este trabajo está sujeto a la Política de acceso público de los NIH.
intereses contrapuestos
Los autores declaran no hay intereses contrapuestos.
Supplementary data
para ver los datos complementarios que acompañan a este artículo, visite el sitio web de la revista en: www.,future-science.com/doi/suppl/10.2144/000114196