Suppression et remplacement précis des gènes à l’aide du système CRISPR/Cas9 dans les cellules humaines
Nous montrons ici que deux ARN guides couplés à Cas9 génèrent efficacement des délétions D’ADN allant jusqu’à 10 kb dans les cellules humaines dans un processus où la réparation de la délétion est De plus, nous fournissons des données montrant que le système CRISPR/Cas9 peut remplacer de gros fragments génomiques en présence d’un donneur de réparation homologue linéaire.,
Les répétitions palindromiques courtes et entrecoupées de bactéries/LOCI associés à CRISPR (CRISPR / Cas) codent des systèmes immunitaires guidés par L’ARN qui protègent les cellules contre les virus et les plasmides envahissants (1, 2). Chez Streptococcus pyogenes, les systèmes CRISPR/Cas de type II utilisent une endonucléase guidée par L’ARN (RGEN), Cas9, pour catalyser le clivage spécifique du site des séquences D’ADN cibles., Le ciblage de Cas9 vers des sites génomiques spécifiques est médié par une séquence guide de 20 nucléotides à l’intérieur d’un ARN CRISPR associé (arnrcr) et nécessite un arnrcr transactif (arnrcr) qui recrute l’arnrcr dans le complexe Cas9 (3). La reconnaissance des sites de clivage est déterminée par l’appariement de bases d’ADN-ARNr et un motif protospacer-adjacent (PAM), une séquence de trois nucléotides (NGG) juxtaposée à la région complémentaire D’ADN (4)., Il est à noter qu’un ARN guide unique (ARNg) imitant le complexe tracrRNA-arncr peut recruter Cas9 sur des sites génomiques ciblés et générer des cassures double brin (DSB) dans L’ADN (5). Les systèmes CRISPR/Cas9 ont été adaptés pour l’édition du génome spécifique à un site dans divers types de cellules et organismes (6-12).
l’édition du génome avec CRISPR / Cas9 est initiée avec l’introduction d’un DSB à un locus génomique ciblé en utilisant le RGEN programmé par ARN. Ceci est suivi par la réparation du DSB par la réparation dirigée par homologie (HDR) ou la jonction d’extrémité non homologue (NHEJ)., En présence d’un donneur de réparation homologue, le système CRISPR/Cas9 peut être utilisé pour générer des modifications et des insertions précises et définies à un lieu ciblé à travers le processus HDR. En l’absence d’un donneur de réparation homologue, les DSB uniques générés par CRISPR/Cas9 sont réparés par le biais du NHEJ sujet aux erreurs, ce qui entraîne des mutations d’insertion ou de délétion (indel). Les mutations Indel dans les exons codants peuvent introduire des codons d’arrêt prématurés ou des mutations de décalage de trame, inactivant ainsi les protéines correspondantes., Les mutations Indel générées par la réparation d’un seul DSB peuvent ne pas être utiles dans des expériences visant à caractériser les domaines fonctionnels de gènes codant des protéines ou pour l’inactivation d’éléments génomiques tels que des séquences régulatrices intergéniques ou introniques ou des gènes D’ARN non codants. Les suppressions de fragments D’ADN dans les loci cibles fourniraient une avenue pour étudier ces éléments fonctionnels. À cette fin, plusieurs DSB ont été introduits pour générer des délétions chez la drosophile (12, 13), le poisson zèbre (14) et les cellules humaines (8), mais avec une faible efficacité., Des délétions ciblées d’ADN génomique ont également été réalisées en utilisant la nucléase à doigts de zinc (ZFN) ou la nucléase effectrice de type activateur de transcription (TALEN) dans des cellules humaines (15-17). Cependant, l’efficacité de ces approches est généralement faible. En outre, ZFNs et TALENs restent quelque peu difficiles et coûteux à concevoir, développer et tester empiriquement dans le contexte cellulaire.
ici, nous avons examiné la génération de délétions de fragments dans des cellules humaines catalysées par le système CRISPR/Cas9. Nous montrons que 2 Grna couplés à Cas9 peuvent efficacement créer des suppressions D’ADN allant jusqu’à 10 kb., D’intérêt, nous avons constaté que la réparation de ce processus de suppression est en grande partie accomplie par l’assemblage final précis. De plus, la suppression ciblée avec CRISPR/Cas9 semble indépendante du statut transcriptionnel du locus ciblé. Enfin, nous montrons que le système CRISPR / Cas9 peut être utilisé pour remplacer de gros fragments génomiques en présence d’un donneur de réparation homologue linéaire.
matériel et méthodes
construction du plasmide
le promoteur H1 de base a été amplifié à partir du plasmide pLVTHM (Addgene, #12247, Cambridge, MA)., Des oligonucléotides contenant le promoteur H1 modifié et l’épine dorsale des séquences d’ARNg désirées avec deux sites BsaI ont été synthétisés (installation PAN, Université Stanford). Les produits de pleine longueur résultants ont été amplifiés par PCR et clonés dans le vecteur pUC19. Le gène ampicilline (amp) et le promoteur H1 du vecteur pUC19 contiennent des sites enzymatiques de restriction BsaI; ceux-ci ont été mutés (le gène amp a été changé de G1601C, ce qui ne modifie pas la séquence d’acides aminés; le promoteur H1 a été changé de GAGACC à GAGGACC) pour éliminer les sites BsaI., Le protocole relatif au clonage des ARNg est présenté dans les documents complémentaires. Toutes les séquences de sites de ciblage sont présentées dans le tableau supplémentaire S1.
culture cellulaire
Les cellules HEK 293T, SK-Hep1 et HeLa ont été mises en culture dans le milieu de Dulbecco »s modified Eagle »s (DMEM) additionné de 10% de sérum fœtal bovin (FBS) (Hyclone, Logan, UT) et de pénicilline/streptomycine (pen / strep) (Invitrogen, Carlsbad, CA). Les cellules PC3 ont été mises en culture dans un milieu RPMI-1640 additionné de 10% de FBS et de pen/strep., Pour la stimulation du facteur de nécrose tumorale α (TNF-α), les cellules 293T ont été traitées avec des concentrations indiquées de TNF-α(R&d Systems, Minneapolis, MN). Les cellules ont été maintenues à 37°C et 5% de CO2 dans un incubateur humidifié.
délétion ciblée de L’ADN
Les cellules HEK 293T ont été ensemencées dans des plaques de 12 puits à une densité de 100 000 cellules par puits. Après 24 h, les cellules ont été transitoirement transfectées avec 1 µg de plasmide Cas9 (Addgene, # 41815), 0,5 µg d’ARNg T1 et 0,5 µg d’ARNg T2 en utilisant Lipofectamine 2000 (Invitrogen) selon les protocoles du fabricant., L’ADN génomique a été extrait 48 h après la transfection à L’aide D’une Solution D’Extraction D’ADN QuickExtract (Epicentre Biotechnologies, Madison, WI). Une PCR commune a été réalisée pour amplifier la région ciblée à l’aide d’amorces flanquant les régions ciblées. Les fragments génomiques de type sauvage et tronqués ont été résolus par électrophorèse sur gel. La PCR en temps réel (RT-PCR) a été réalisée pour quantifier le pourcentage de suppression à l’aide d’amorces à travers la jonction ou dans la région de suppression. La méthode CQ comparative a été utilisée pour calculer le niveau d’expression de la région cible par rapport à une région de référence (locus ACTB)., Le pourcentage de délétion dans la région cible a été calculé en outre par le rapport des cellules cibles par rapport aux cellules témoins. Toutes les séquences d’amorces sont énumérées dans le tableau supplémentaire S2.
séquençage cible
Les cellules ont été récoltées deux jours après la transfection, et l’ADN génomique a été extrait à L’aide D’une Solution D’Extraction D’ADN à extraction rapide (Epicentre Biotechnologies). La PCR a été menée pour amplifier la région de ciblage avec de l’ADN génomique dérivé des cellules, et les amplicons ont été séquencés en profondeur par le séquenceur personnel MiSeq (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Les séquences de donneurs sont présentées dans le matériel supplémentaire. Les deux liaisons les plus 5‘sont le phosphorothioate (indiqué par des astérisques). Les cellules des plaques à 6 puits ont été transitoirement transfectées avec 2,0 µg de plasmide Cas9, 0,8 µg de plasmide T1 d’ARNg, 0,8 µg de plasmide T2 d’ARNg et 0,4 µg de donneur linéaire à L’aide de Lipofectamine 2000 (Invitrogène). À 48 h après la transfection, les cellules ont été traitées avec 1 ng/mL de TNF-α pendant 24 h, puis les cellules GFP-positives ont été triées.
analyse de la luciférase
Pour l’analyse de la luciférase, les cellules HEK 293T ont été ensemencées dans des plaques de 96 puits à une densité de 5000 cellules par puits., Après 24 h, les cellules ont été transitoirement transfectées avec 5 ng de PRL-TK Renilla luciferase reporter et 100 ng de luciferase reporter avec cytomégalovirus (CMV), SV40 (virus Simien 40) ou promoteur basique. Après 48 h, l’activité de la luciférase a été mesurée avec le double système d’analyse de la luciférase reporter (Promega).
Western blot
Les protéines ont été séparées par le dodécylsulfate de sodium—PAGE (SDS-PAGE) et transférées aux membranes de nitrocellulose. Les membranes ont été bloquées avec 5% de lait non gras et incubées avec un anticorps GFP (CST, #2555S, Danvers, MA)., Le complexe antigène-anticorps a été détecté avec des réactifs de chimiluminescence améliorés.
résultats et discussion
Nous avons adapté le système bactérien CRISPR / Cas9 de type II pour mutagéniser L’ADN génomique dans les cellules humaines. La version optimisée pour le codon humain de la protéine S. pyogenes Cas9 portant un signal de localisation nucléaire C-terminus SV40 a été exprimée à l’aide d’un système décrit précédemment (6). Pour diriger le clivage Cas9 vers la séquence souhaitée, nous avons exprimé des transcriptions de fusion crRNA-tracrRNA, ci-après dénommées ARN guide (ARN), à partir d’un promoteur H1 polymérase III humain modifié., L’extrémité 3‘ du promoteur H1 a été modifiée pour permettre la transcription des ARN qui commencent par n’importe quel nucléotide. Contrainte seulement par l’exigence que la cible de l’ARNr de 20 bp soit suivie de la séquence PAM, NGG (où N est n’importe quel nucléotide), cette approche peut en principe être utilisée pour cibler n’importe quel emplacement génomique qui a la forme N20NGG. Pour faciliter le clonage du vecteur d’expression gRNA, nous avons utilisé une enzyme de restriction de type IIs, BsaI. Cela a nécessité la synthèse d’un oligonucléotide de 24 bp contenant une région de complémentarité au site cible sur L’ADN., Le protocole simple et efficace pour le clonage du vecteur d’expression gRNA (Figure 1a) est décrit en détail dans le matériel supplémentaire.
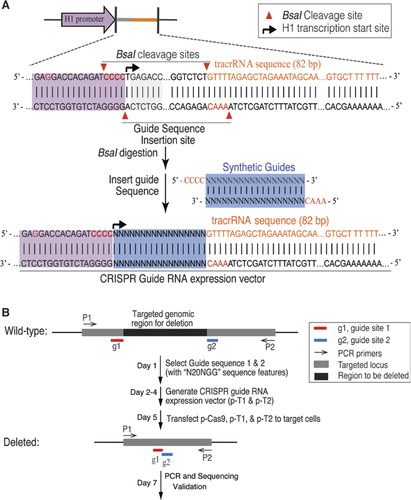
(a) conception du vecteur d’expression de l’ARN guide (gRNA). Le vecteur a été conçu pour produire des transcriptions d’ARNg avec un ARNg synthétique fusionné à L’ARN trans-activant/ARN CRISPR (tracrRNA)., Le promoteur H1 a été modifié pour éliminer le site interne de l’enzyme de restriction BSAI de type IIS en changeant GAGACC dans le promoteur H1 en GAGGACC. Un site BsaI a été introduit pour créer des sites de Clonage pour les fusions gRNA et tracrRNA par insertion de duplexes oligonucléotides synthétiques avec des surplombs compatibles. L’extrémité 3‘ du promoteur H1 a été modifiée pour être CCACAGATCCCC afin de faciliter la transcription des ARNr avec n’importe quel nucléotide à l’extrémité 5‘. B) les étapes de la délétion ciblée du gène avec CRISPR/Cas9.,
pour la délétion d’un grand segment d’ADN génomique, nous avons utilisé une paire d’ARNr contre le locus ciblé (Figure 1B). Deux sites cibles avec le motif N20NGG ont été sélectionnés à la limite de la région cible. L’efficacité de la délétion ciblée guidée par diverses combinaisons de paires d’ARNg a été déterminée par des analyses PCR utilisant des amorces flanquant les régions ciblées. Les fragments génomiques de type sauvage et tronqués ont été résolus par électrophorèse sur gel. Pour éviter le biais D’amplification par PCR, le pourcentage de suppression a été quantifié par RT-PCR à l’aide d’une paire d’amorces., Les amorces ont été conçues à travers la jonction de suppression (une amorce en dehors de la région de suppression, l’autre amorce dans la région de suppression) ou dans les régions de suppression (les deux amorces se trouvent dans la région de suppression). Ainsi, une seule bande est amplifiée avec la paire d’amorces pour les cellules ciblées et les cellules de contrôle. Nous avons calculé le pourcentage de délétion en comparant la quantité relative de produits PCR (cellules cibles par rapport aux cellules témoins) amplifiés par la même paire d’amorces. Les suppressions ciblées ont été vérifiées par séquençage.,
pour évaluer comment les paires d’ARNg pourraient affecter la réparation ultérieure et la génération de suppressions, nous avons d’abord conçu des ensembles d’ARNg ciblés contre le locus génomique humain CDC42 et séparés par des distances allant d’environ 200 à 10 000 PB (Figure 2a et tableau supplémentaire S1). Nous avons ensuite évalué la capacité de chaque paire d’ARNg à générer des délétions dans des cellules HEK 293T humaines en présence de Cas9. L’efficacité robuste des suppressions basées sur le NHEJ (jusqu’à 68%) a été confirmée par le qPCR (Figure 2B-2D)., Même pour la délétion d’une région génomique de 10 kb, nous avons obtenu des taux de ciblage de 16% à 28%, selon la paire d’ARNg. Ce processus d’édition médié par L’ARN a été rapide, la première délétion détectable apparaissant environ 12 h après la transfection (figure supplémentaire S1). Le système a été efficace dans une variété de types cellulaires, y compris: les cellules PC3, SK-Hep1 et hela (figure supplémentaire S2).

(a) schéma représentant les emplacements des ARN guides (ARN) ciblant le locus CDC42. (B-D) L’efficacité de la délétion ciblée avec CRISPR/Cas9 a été déterminée par PCR dans des cellules HEK 293T. Des amorces en dehors des régions de suppression attendues ont été utilisées. Le pourcentage de suppression a été quantifié par RT-PCR en utilisant des amorces à travers la jonction ou dans la région de suppression. E–F) l’efficacité et la précision de la suppression ciblée dans CDC42 ont été confirmées par E) le séquençage Sanger et F) les analyses de séquençage à haut débit. Le produit PCR contenant uniquement l’amplicon de délétion a été enrichi pour le séquençage.,
les délétions ont été confirmées par séquençage de produits PCR couvrant les sites de clivage attendus. Le séquençage de Sanger a montré que les jonctions de délétion résultaient de la ligature précise des DSB à extrémité émoussée créées par Cas9; chaque DSB se produisait exactement 3 PB en amont de la séquence PAM (Figure 2e et figure supplémentaire S3). Nous avons également utilisé le séquençage profond des amplicons de suppression pour évaluer la précision de l’efficacité de la suppression; dans environ 80% des lectures, les DSB ciblés ont été parfaitement réparés (Figure 2F).,
Nous avons récapitulé ces résultats en testant des paires d’ARNg conçues pour supprimer des fragments d’un locus génomique contenant le gène microARN mir-21 dans des cellules HEK 293T. Deux ARN ont été conçus pour cibler les limites de l’épingle à cheveux miR-21 (figure supplémentaire S4). L’efficacité de délétion a été de 38% après transfection avec les deux ARN et Cas9, mesurée à l’aide d’un test PCR (figure supplémentaire S4B). Le séquençage profond a confirmé que la suppression s’est produite exactement comme prévu (figure supplémentaire S4C).,
pour déterminer si le système de délétion des gènes médié par CRISPR / Cas9 est influencé par l’état transcriptionnel des gènes ciblés, le gène codant pour le ligand 2 (CCL2) de la chimiokine (motif C-C) a été ciblé. CCL2 est une petite cytokine appartenant à la famille des chimiokines CC; le gène CCL2 est une cible de la signalisation NF-kB. Nous avons sélectionné au hasard huit sites cibles situés aux extrémités 5‘ et 3 ‘ du locus du gène CCL2 (Figure 3A). Nous avons obtenu une délétion robuste et efficace de différentes régions du gène en utilisant Cas9 et différentes paires d’ARNr dans les cellules HEK 293T (Figure 3B–3D).,

(a) schéma représentant les emplacements des ARN guides (ARN) ciblant le locus CCL2. (B–D) l’efficacité de la délétion ciblée de CCL2 avec CRISPR/Cas9 dans les cellules HEK 293T. (E) les niveaux D’ARNm CCL2 ont été déterminés par addition du facteur de nécrose tumorale α (TNF-α) par des analyses quantitatives RT-PCR dans des cellules HEK 293T. Les données ont été présentées avec les moyennes ± sem en trois exemplaires., (F) l’efficacité de la délétion ciblée de CCL2 avec CRISPR/Cas9 après traitement par TNF-α pendant 24 h dans les cellules HEK 293T. (G) le niveau d’activité luciférase du cytomégalovirus (CMV), ou SV40, ou d’un promoteur basique dans les cellules HEK 293T. Les données ont été présentées avec les moyennes ± sem en trois exemplaires. (H) efficacité de la délétion ciblée d’un gène de la luciférase contrôlé par le CMV, ou SV40, ou un promoteur basique dans les cellules HEK 293T. (B-D, F, H) le pourcentage de délétion a été quantifié par RT-PCR en utilisant des amorces à travers la jonction ou dans la région de délétion.,
L’Expression du gène CCL2 a été induite de façon spectaculaire par le TNF-α (jusqu’à increase 300 fois plus) dans les cellules HEK 293T (Figure 3E), fournissant un bon modèle pour étudier si L’édition ciblée du génome médiée par CRISPR / Cas9 est affectée par l’activité transcriptionnelle. Il est intéressant de noter que l’efficacité de la délétion ciblée du locus du gène CCL2 n’a pas été affectée par le traitement des cellules avec le TNF-α(Figure 3F), ce qui suggère que la transcription n’a pas altéré la délétion médiée par CRISPR/Cas9., Pour confirmer davantage ce résultat, nous avons ciblé un gène rapporteur exogène piloté par divers promoteurs avec des forces différentes, où les différentes activités transcriptionnelles pourraient être évaluées à l’aide d’un test de luciférase (Figure 3G). Les tests de PCR ont révélé une efficacité similaire dans les délétions ciblées dans les cellules HEK 293T après co-transfection du gène rapporteur avec des paires Cas9 et gRNA (Figure 3H). Ce résultat indique que la réparation induite par la NHEJ peut se produire malgré l’apparition de divers degrés d’activité transcriptionnelle.,
Les DSBs peuvent stimuler le HDR pour permettre un remplacement très précis de la région endommagée par un donneur homologue. Pour obtenir un remplacement ciblé de L’ADN génomique, nous avons introduit une paire d’ARN, Cas9, et un donneur linéaire avec homologie à la région ciblée dans les cellules (Figure 4A). Le donneur linéaire a été obtenu par amplification PCR avec des amorces portant une séquence homologue de 50 PB. Ce même donneur a été inséré avec succès à l’aide d’un système de réparation HDR basé sur ZFN (18)., Pour tester la faisabilité D’un remplacement médié par CRISPR/ Cas9 par HDR, nous avons ciblé le locus CCL2 avec une paire de Grna (#39 et # 1854 illustrés à la Figure 3) et un donneur portant la séquence codante de la protéine fluorescente verte améliorée (EGFP) et le site SV40 poly(A) (Figure 4A; les séquences et les positions sont présentées dans le matériel supplémentaire). En utilisant ce système, environ 0,5% des cellules ciblées étaient positives à L’EGFP, alors que seulement 0,023% étaient positives à L’EGFP dans les cellules de transfection simulées (juste transfectées avec un donneur), ce qui était similaire aux cellules témoins (0,021%, sans transfection)., Les cellules EGFP-positives ont ensuite été triées par cytométrie en flux. L’intégration spécifique au Site a été confirmée par PCR à l’aide de deux paires d’amorces flanquant à la fois les bras homologues et toute la région remplacée. Comme le montre la Figure 4B, nous avons observé la région remplacée attendue contenant la séquence EGFP pleine longueur et les bras homologues (résultat du séquençage de Sanger montré dans le matériel supplémentaire). L’allèle endogène de type sauvage a également été détecté (Figure 4B), ce qui indique que tous les allèles ne sont pas ciblés., De plus, nous avons sélectionné des clones uniques parmi les cellules EGFP-positives et avons constaté que tous les clones (6 sur 6 examinés) avaient l’intégration attendue (Figure 4C), mais l’allèle endogène de type sauvage a également été détecté dans trois des clones (Figure 4c), suggérant qu’un seul allèle était ciblé dans ces clones. L’Expression de la protéine EGFP dans les cellules ciblées (cellules triées positives EGFP) a été régulée à la hausse sur le traitement TNF-α tel qu’évalué par Western blot et fluorescence activated cell sorting (FACS) (Figure 4, D et E)., Ces résultats ont démontré que le système CRISPR / Cas9 peut être utilisé pour créer des remplacements de gènes/domaines avec une efficacité et une précision élevées.

(a) diagrammes schématiques illustrant la procédure de remplacement ciblé de gènes à L’aide de CRISPR/Cas9 dans des cellules humaines., Pour tester l’efficacité du remplacement ciblé du gène, des ARN guides (ARN) ont été conçus pour supprimer la région indiquée (marquée par les sites 1 et 2) du gène CCL2 et pour remplacer la région supprimée par le donneur de cassette EGFP-polyA avec des bras avec de courtes régions d’homologie. Les sites ciblés du site 1 et du site 2 sont #39 et #1854 dans le gène CCL2 illustré à la Figure 3. Les séquences homologues (50 PB) sont juste en amont et en aval des sites de délétion. Les positions et les séquences en détail sont présentées dans le matériel supplémentaire., B) L’efficacité du remplacement ciblé des gènes par CRISPR/Cas9 a été déterminée par PCR dans des cellules HEK 293T. Des amorces couvrant les jonctions entre CCL2 et EGFP ont été utilisées pour l’amplification par PCR. C) analyse par PCR pour le remplacement ciblé des gènes des clones individuels. (D,E) L’Expression de la protéine EGFP lors de L’addition de TNF-α dans les cellules HEK 293T a été déterminée par (D) western blot et (E) FACS.
Nous décrivons ici une approche simple et efficace pour la délétion de gènes en utilisant le système CRISPR / Cas9., Nous avons démontré que l’introduction de ce système dans des cellules HEK 293T humaines, et d’autres types de cellules humaines, induisait des délétions de fragments jusqu’à 10 kb avec des efficacités comprises entre 11% et 68%, selon la séquence ciblée. La capacité de supprimer efficacement et précisément les segments génomiques facilitera l’étude des éléments génomiques fonctionnels dans les cellules humaines. Cette approche peut potentiellement être utilisée pour cibler n’importe quel loci génomique.
on s’est inquiété de la spécificité du système CRISPR/Cas9 (19-21)., Pour exclure les phénotypes indésirables dus à des mutations hors cible, nous suggérons d’utiliser au moins deux paires d’ARNr différentes pour chaque région cible. Dans notre étude, le besoin de plusieurs paires d’ARNg n’était pas une limitation majeure, étant donné la simplicité et la grande efficacité de ce système. Il est à noter que différentes paires d’ARN ciblant la même région ont fonctionné avec une grande efficacité (Figures 2 et 4). Une autre approche pour éviter les mutations indésirables est l’utilisation de la méthode de la Double nickase (22, 23)., Nous avons également appliqué avec succès la méthode de la Double nickase pour générer la délétion de l’ADN génomique, mais l’efficacité était considérablement inférieure.
Il est connu que la réparation des DSBs D’ADN est largement médiée par la NHEJ sujette aux erreurs, dans laquelle les deux extrémités sont traitées et ligaturées ensemble d’une manière qui s’accompagne fréquemment d’insertions et de délétions de nucléotides. Une telle jonction d’extrémité sujette aux erreurs a été observée dans la réparation des DSB créés par ZFNs ou TALENs. En revanche, la réparation des DSB générés par Cas9 et deux Grna a été très précise., Nos résultats suggèrent que les ruptures sont directement ligaturées sans traitement final, révélant un avantage auparavant méconnu de la voie NHEJ. Le mécanisme qui aboutit aux ligatures précises reste à déterminer. Une possibilité est que la suppression ciblée en utilisant Cas9 et deux grnas entraîne une jonction qui n’est reconnue par aucun des gRNAs d’origine. Nous avons également analysé l’efficacité dans la génération de mutations indel pour la paire individuelle gRNA et gRNA (#39 et #224 gRNA sur la Figure 3a) par séquençage Sanger des amplicons PCR (Clonage TA)., D’intérêt, nous avons observé que l’efficacité de la génération de mutations indel pour l’ARNg unique était assez faible (9,5%, 2 clones sur 21 pour l’ARNg #39; 5%, 1 clone sur 20 pour l’ARNg #224). Cependant, la paire gRNA a généré une efficacité élevée des mutations indel (50%, 10 des 20 clones pour #39 et #224), ce qui était similaire à l’analyse utilisant qPCR (52%, Figure 3B). Nous proposons qu’un seul gRNA entraîne souvent une extrémité émoussée du site de clivage, qui sera précisément réparée par NHEJ. Ainsi, l’efficacité de générer une mutation est beaucoup plus faible avec l’utilisation d’un seul ARNg qu’une paire d’ARNg.,
Remerciements
ce travail a été soutenu par des subventions de L’Institut national de la santé (No. DP1CA174421) et la fondation W. M. Keck à C.-Z. C, et la Fondation Nationale des sciences naturelles de Chine (No.81101481) et le programme de formation des talents médicaux de Shanghai (No. XYQ2011048) à S. L. H. ce document est soumis à la Politique D’accès Public des NIH.
intérêts divergents
Les auteurs déclarent n’avoir aucun conflit d’intérêts.
données Supplémentaires
Pour afficher les données supplémentaires qui accompagnent ce document, veuillez visiter le journal site internet: www.,future-science.com/doi/suppl/10.2144/000114196