Delezione e sostituzione precise del gene facendo uso del sistema CRISPR / Cas9 in cellule umane
Qui mostriamo che due RNA della guida accoppiati con Cas9 generano efficientemente le eliminazioni del DNA di fino a 10 kb in cellule umane in un processo in cui la riparazione della delezione è in gran parte compiuta Inoltre, forniamo dati che dimostrano che il sistema CRISPR/Cas9 può sostituire grandi frammenti genomici in presenza di un donatore di riparazione omologo lineare.,
Il clustered batterica regolarmente interspaced brevi ripetizioni palindromiche / CRISPR—associated (CRISPR / Cas) loci codificano sistemi immunitari RNA-guidati che proteggono le cellule contro virus invasori e plasmidi (1, 2). Nello Streptococcus pyogenes, i sistemi CRISPR/Cas di tipo II utilizzano un’endonucleasi guidata da RNA (RGEN), Cas9, per catalizzare la scissione site-specific delle sequenze di DNA bersaglio., Il targeting di Cas9 a specifici siti genomici è mediato da una sequenza guida di 20 nucleotidi all’interno di un RNA CRISPR associato (crRNA) e richiede un crRNA transattivante (tracrRNA) che recluta il crRNA nel complesso Cas9 (3). Il riconoscimento dei siti di scissione è determinato dall’accoppiamento base crRNA-DNA e da un motivo protospacer-adjacent (PAM), una sequenza di tre nucleotidi (NGG) giustapposta alla regione complementare del DNA (4)., È notevole che un singolo RNA guida (gRNA) che imita il complesso tracrRNA-crRNA può reclutare Cas9 in siti genomici mirati e generare rotture a doppio filamento (DSB) nel DNA (5). I sistemi CRISPR / Cas9 sono stati adattati per l’editing del genoma sito-specifico in diversi tipi di cellule e organismi (6-12).
La modifica del genoma con CRISPR / Cas9 viene avviata con l’introduzione di un DSB in un locus genomico mirato utilizzando il RGEN programmato dall’RNA. Questa è seguita dalla riparazione del DSB attraverso la riparazione diretta all’omologia (HDR) o l’unione finale non omologica (NHEJ)., In presenza di un donatore di riparazione omologa, il sistema CRISPR/Cas9 può essere utilizzato per generare modifiche e inserimenti precisi e definiti in un luogo mirato attraverso il processo HDR. In assenza di un donatore di riparazione omologa, i singoli DSB generati da CRISPR/Cas9 vengono riparati attraverso il NHEJ soggetto a errori, che si traduce in mutazioni di inserimento o delezione (indel). Le mutazioni Indel negli esoni codificanti possono introdurre codoni stop prematuri o mutazioni frame-shift, inattivando così le proteine corrispondenti., Le mutazioni Indel generate dalla riparazione di un singolo DSB potrebbero non essere utili in esperimenti volti a caratterizzare i domini funzionali dei geni codificanti proteine o per l’inattivazione di elementi genomici come sequenze regolatorie intergeniche o introniche o geni di RNA non codificanti. Le eliminazioni di frammenti di DNA nei loci bersaglio fornirebbero una via per studiare questi elementi funzionali. A tal fine, sono stati introdotti più DSB per generare eliminazioni in Drosophila (12, 13), zebrafish (14) e cellule umane (8), anche se con bassa efficienza., Le eliminazioni mirate del DNA genomico inoltre sono state raggiunte facendo uso della nucleasi del dito dello zinco (ZFN) o della nucleasi effettrice del tipo dell’attivatore della trascrizione (TALEN) in cellule umane (15-17). Tuttavia, le efficienze di questi approcci sono generalmente basse. Inoltre, ZFNs e TALENs rimangono alquanto difficili e costosi da progettare, sviluppare e testare empiricamente nel contesto cellulare.
Qui, abbiamo esaminato la generazione di eliminazioni di frammenti in cellule umane catalizzate dal sistema CRISPR/Cas9. Dimostriamo che 2 GRNA accoppiati con Cas9 possono creare in modo efficiente eliminazioni di DNA fino a 10 kb., Di interesse, abbiamo scoperto che la riparazione di questo processo di eliminazione è in gran parte compiuta da fine preciso unendo. Inoltre, la delezione mirata con CRISPR/Cas9 sembra essere indipendente dallo stato trascrizionale del locus mirato. Infine, mostriamo che il sistema CRISPR / Cas9 può essere utilizzato per sostituire grandi frammenti genomici in presenza di un donatore di riparazione omologo lineare.
Materiale e metodi
Costruzione plasmidica
Il promotore H1 di base è stato amplificato da PLVTHM plasmid (Addgene, #12247, Cambridge, MA)., Sono stati sintetizzati oligonucleotidi contenenti il promotore H1 modificato e la spina dorsale delle sequenze GRNA desiderate con due siti BsaI (PAN Facility, Stanford University). I prodotti a lunghezza intera risultanti sono stati amplificati mediante PCR e clonati nel vettore pUC19. Il gene ampicillina (amp) e il promotore H1 nel vettore pUC19 contengono siti enzimatici di restrizione BsaI; questi sono stati mutati (il gene amp è stato cambiato da G1601C, che non altera la sequenza aminoacidica; il promotore H1 è stato cambiato da GAGACC a GAGGACC) per eliminare i siti BsaI., Il protocollo per la clonazione di gRNA è presentato nel Materiale supplementare. Tutte le sequenze di siti di targeting sono presentate nella tabella supplementare S1.
Coltura cellulare
Le cellule HEK 293T, SK-Hep1 e HeLa sono state coltivate nel mezzo di Eagle modificato di Dulbecco (DMEM) integrato con siero bovino fetale al 10% (FBS) (Hyclone, Logan, UT) e penicillina/streptomicina (pen / strep) (Invitrogen, Carlsbad, CA). Le cellule PC3 sono state coltivate nel mezzo RPMI-1640 integrato con 10% FBS e pen/strep., Per la stimolazione del fattore di necrosi tumorale α (TNF-α), le cellule 293T sono state trattate con concentrazioni indicate di TNF-α(R&D Systems, Minneapolis, MN). Le cellule sono state mantenute a 37°C e al 5% di CO2 in un incubatore umidificato.
Eliminazione mirata del DNA
Le cellule HEK 293T sono state seminate in piastre da 12 pozzetti ad una densità di 100.000 cellule per pozzetto. Dopo 24 ore, le cellule sono state transitoriamente trasfettate con plasmide 1 µg Cas9 (Addgene, #41815), 0,5 µg gRNA T1 e 0,5 µg GRNA T2 plasmidi utilizzando Lipofectamine 2000 (Invitrogen) secondo i protocolli del produttore., Il DNA genomico è stato estratto 48 h dopo la trasfezione utilizzando la soluzione di estrazione del DNA QuickExtract (Epicentre Biotechnologies, Madison, WI). La PCR comune è stata condotta per amplificare la regione mirata utilizzando primer che fiancheggiano le regioni mirate. I frammenti genomici di tipo selvaggio e troncati sono stati risolti mediante elettroforesi su gel. La PCR in tempo reale (RT-PCR) è stata eseguita per quantificare la percentuale di eliminazione utilizzando primer attraverso la giunzione o all’interno della regione di eliminazione. Il metodo Cq comparativo è stato utilizzato per calcolare il livello di espressione della regione target rispetto a una regione di riferimento (locus ACTB)., La percentuale di delezione nella regione target è stata ulteriormente calcolata dal rapporto tra le cellule target rispetto alle cellule di controllo. Tutte le sequenze di primer sono elencate nella Tabella supplementare S2.
Sequenziamento target
Le cellule sono state raccolte due giorni dopo la trasfezione e il DNA genomico è stato estratto utilizzando la soluzione di estrazione del DNA QuickExtract (Epicentre Biotechnologies). La PCR è stata condotta per amplificare la regione di targeting con DNA genomico derivato dalle cellule e gli ampliconi sono stati sequenziati in profondità da Miseq Personal Sequencer (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Le sequenze donatrici sono presentate nel Materiale supplementare. I due 5 ‘ – la maggior parte dei collegamenti sono fosforotioato (indicato da asterischi). Le cellule nelle piastre a 6 pozzetti sono state transitoriamente trasfettate con 2,0 µg di plasmide Cas9, 0,8 µg di plasmide gRNA T1, 0,8 µg di plasmide gRNA T2 e 0,4 µg di donatore lineare utilizzando Lipofectamina 2000 (Invitrogen). A 48 h dopo la trasfezione, le cellule sono state trattate con 1 ng / mL TNF-α per 24 h, e quindi le cellule GFP-positive sono state ordinate.
Saggio di luciferasi
Per il saggio di luciferasi, le cellule HEK 293T sono state seminate in piastre a 96 pozzetti ad una densità di 5000 cellule per pozzetto., Dopo 24 ore, le cellule sono state transitoriamente trasfettate con 5 ng di PRL-TK Renilla luciferasi reporter e 100 ng di luciferasi reporter con citomegalovirus (CMV), SV40 (Simian virus 40) o promotore di base. Dopo 48 ore, l’attività della luciferasi è stata misurata con il dual luciferase reporter assay system (Promega).
Western blot
Le proteine sono state separate dalla PAGINA di dodecil solfato di sodio (SDS—PAGE) e trasferite alle membrane di nitrocellulosa. Le membrane sono state bloccate con latte non grasso al 5% e incubate con anticorpi GFP (CST, #2555S, Danvers, MA)., Il complesso antigene-anticorpo è stato rilevato con reagenti di chemiluminescenza potenziati.
Risultati e discussione
Abbiamo adattato il sistema batterico CRISPR / Cas9 di tipo II per mutagenizzare il DNA genomico nelle cellule umane. La versione ottimizzata per il codone umano della proteina S. pyogenes Cas9, con un segnale di localizzazione nucleare C-terminus SV40, è stata espressa utilizzando un sistema precedentemente descritto (6). Per indirizzare la scissione Cas9 alla sequenza desiderata, abbiamo espresso i trascritti di fusione crRNA-tracrRNA, di seguito denominati RNAS guida (gRNAs), da un promotore umano H1 polimerasi III modificato., L’estremità 3‘ del promotore H1 è stata modificata per consentire la trascrizione di GRNA che iniziano con qualsiasi nucleotide. Vincolato solo dal requisito che il bersaglio CRRNA 20 bp essere seguito dalla sequenza PAM, NGG (dove N è qualsiasi nucleotide), questo approccio può in linea di principio essere utilizzato per indirizzare qualsiasi posizione genomica che ha la forma N20NGG. Per facilitare la clonazione del vettore di espressione gRNA, abbiamo usato un enzima di restrizione di tipo IIs, BsaI. Ciò ha richiesto la sintesi di un oligonucleotide di 24 bp contenente una regione di complementarità al sito bersaglio sul DNA., Il protocollo semplice ed efficiente per la clonazione del vettore di espressione gRNA (Figura 1A) è descritto in dettaglio nel Materiale supplementare.
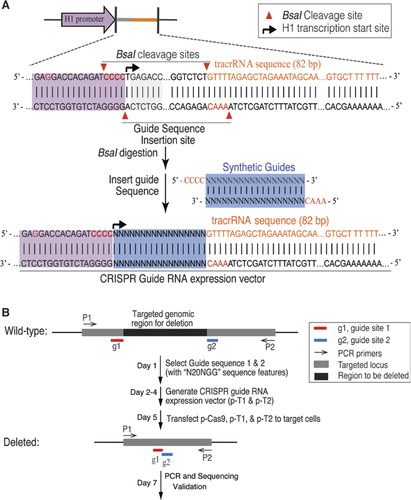
(A) Disegno del vettore di espressione dell’RNA guida (gRNA). Il vettore è stato progettato per produrre trascritti di gRNA con un gRNA sintetico fuso all’RNA transattivante/RNA CRISPR (tracrRNA)., Il promotore H1 è stato modificato per eliminare il sito interno dell’enzima di restrizione di tipo IIS BsaI cambiando GAGACC nel promotore H1 in GAGGACC. È stato introdotto un sito BsaI per creare siti di clonazione per fusioni gRNA e tracrRNA mediante inserimento di duplex oligonucleotidici sintetici con sporgenze compatibili. L’estremità 3‘ del promotore H1 è stata modificata per essere CCACAGATCCCC per facilitare la trascrizione di GRNA con qualsiasi nucleotide all’estremità 5′. (B) Le fasi della delezione genica mirata con CRISPR/Cas9.,
Per la cancellazione di un ampio segmento di DNA genomico, abbiamo usato una coppia di GRNA contro il locus mirato (Figura 1B). Due siti di destinazione con il modello N20NGG sono stati selezionati al confine della regione di destinazione. L’efficienza della delezione mirata guidata da varie combinazioni di coppie di gRNA è stata determinata mediante analisi PCR utilizzando primer che fiancheggiano le regioni mirate. I frammenti genomici di tipo selvaggio e troncati sono stati risolti mediante elettroforesi su gel. Per evitare il bias di amplificazione PCR, l’eliminazione percentuale è stata quantificata da RT-PCR utilizzando una coppia di primer., I primer sono stati progettati attraverso la giunzione di cancellazione (un primer al di fuori della regione di cancellazione, l’altro primer all’interno della regione di cancellazione) o all’interno delle regioni di cancellazione (entrambi i primer si trovano all’interno della regione di cancellazione). Pertanto, solo una singola banda viene amplificata con la coppia di primer sia per le cellule mirate che per le cellule di controllo. Abbiamo calcolato l’eliminazione percentuale confrontando la quantità relativa di prodotti PCR (cellule bersaglio contro cellule di controllo) amplificata dalla stessa coppia di primer. Le eliminazioni mirate sono state ulteriormente verificate mediante sequenziamento.,
Per valutare come le coppie di gRNA potrebbero influenzare la successiva riparazione e la generazione di eliminazioni, abbiamo inizialmente progettato set di GRNA mirati al locus genomico CDC42 umano e separati da distanze che vanno da circa 200 a 10.000 bp (Figura 2A e Tabella supplementare S1). Abbiamo quindi valutato la capacità di ciascuna coppia di gRNA di generare eliminazioni in cellule HEK 293T umane in presenza di Cas9. Le forti efficienze delle eliminazioni basate su NHEJ (fino al 68%) sono state confermate da qPCR (Figura 2B–2D)., Anche per la cancellazione di una regione genomica da 10 kb, abbiamo ottenuto tassi di targeting dal 16% al 28%, a seconda della coppia gRNA. Questo processo di editing mediato dall’RNA è stato rapido, con la prima delezione rilevabile che è apparsa circa 12 ore dopo la trasfezione (Figura supplementare S1). Il sistema è stato efficace in una varietà di tipi di cellule, tra cui: PC3, SK-Hep1 e cellule HeLa (figura supplementare S2).

(A) Diagramma schematico raffigurante le posizioni di RNAS guida (gRNAs) targeting locus CDC42. (B-D) L’efficienza della delezione mirata con CRISPR/Cas9 è stata determinata mediante PCR in cellule HEK 293T. Sono stati utilizzati primer al di fuori delle regioni di eliminazione previste. La percentuale di eliminazione è stata quantificata da RT-PCR utilizzando primer attraverso la giunzione o all’interno della regione di eliminazione. (E-F) L’efficienza e la precisione della delezione mirata nel CDC42 sono state confermate da (E) analisi di sequenziamento Sanger e (F) analisi di sequenziamento ad alto throughput. Il prodotto PCR contenente solo l’amplicone di delezione è stato arricchito per il sequenziamento.,
Le eliminazioni sono state ulteriormente confermate dal sequenziamento dei prodotti PCR che coprono i siti di scissione previsti. Il sequenziamento di Sanger ha mostrato che le giunzioni di cancellazione derivano dalla legatura precisa dei DSB smussati creati da Cas9; ogni DSB si è verificato esattamente 3 bp a monte della sequenza PAM (Figura 2E e figura supplementare S3). Abbiamo anche usato il sequenziamento profondo degli ampliconi di delezione per valutare l’accuratezza dell’efficienza di delezione; in circa l ‘ 80% delle letture, i DSB mirati sono stati perfettamente riparati (Figura 2F).,
Abbiamo ricapitolato questi risultati testando coppie gRNA progettate per eliminare frammenti da un locus genomico contenente il gene microRNA miR-21 in cellule HEK 293T. Due GRNA sono stati progettati per colpire i confini della forcella miR-21 (figura supplementare S4). L’efficienza di delezione è stata del 38% dopo la trasfezione con i due GRNA e Cas9, misurata utilizzando un test PCR (figura supplementare S4B). Il sequenziamento profondo ha confermato che la delezione si è verificata esattamente come previsto (Figura supplementare S4C).,
Per indagare se il sistema di delezione genica mediato da CRISPR / Cas9 è influenzato dallo stato trascrizionale dei geni mirati, il gene che codifica il ligando chemochine (C-C motif) 2 (CCL2) è stato mirato. CCL2 è una piccola citochina appartenente alla famiglia delle chemochine CC; il gene CCL2 è un bersaglio della segnalazione NF-kB. Abbiamo selezionato casualmente otto siti target situati nelle estremità 5‘ e 3 ‘ del locus del gene CCL2 (Figura 3A). Abbiamo ottenuto la cancellazione robusta ed efficiente di diverse regioni del gene utilizzando Cas9 e diverse coppie di GRNA in cellule HEK 293T (Figura 3B–3D).,

(A) Diagramma schematico raffigurante le posizioni di RNAS guida (gRNAs) targeting locus CCL2. (B-D) L’efficienza della delezione mirata di CCL2 con CRISPR/Cas9 in cellule HEK 293T. (E) I livelli di mRNA CCL2 sono stati determinati in base all’aggiunta del fattore di necrosi tumorale α (TNF-α) mediante analisi quantitative RT-PCR in cellule HEK 293T. I dati sono stati mostrati con i mezzi ± sem in triplice copia., F) L’efficienza della delezione mirata di CCL2 con CRISPR/Cas9 dopo il trattamento con TNF-α per 24 ore in cellule HEK 293T. (G) Il livello di attività luciferasi da citomegalovirus (CMV), o SV40, o un promotore di base in cellule HEK 293T. I dati sono stati mostrati con i mezzi ± sem in triplice copia. H) Efficacia della delezione mirata di un gene luciferasi controllato da CMV, o SV40, o da un promotore di base in cellule HEK 293T. (B-D, F, H) La percentuale di delezione è stata quantificata da RT-PCR utilizzando primer attraverso la giunzione o all’interno della regione di delezione.,
L’espressione del gene CCL2 è stata drammaticamente indotta dal TNF-α (aumento fino a ∼300 volte) nelle cellule HEK 293T (Figura 3E), fornendo un buon modello per indagare se l’editing mirato del genoma mediato da CRISPR / Cas9 è influenzato dall’attività trascrizionale. Di interesse, l’efficienza della delezione mirata del locus del gene CCL2 non è stata influenzata dal trattamento delle cellule con TNF-α(Figura 3F), suggerendo che la trascrizione non ha alterato la delezione mediata da CRISPR/Cas9., Per confermare ulteriormente questo risultato, abbiamo preso di mira un gene reporter esogeno guidato da vari promotori con diversi punti di forza, in cui le diverse attività trascrizionali potrebbero essere valutate utilizzando un test di luciferasi (Figura 3G). I test PCR hanno rivelato un’efficienza simile nelle eliminazioni mirate nelle cellule HEK 293T dopo la co-trasfezione del gene reporter insieme alle coppie Cas9 e gRNA (Figura 3H). Questo risultato indica che la riparazione mediata da NHEJ può verificarsi nonostante il verificarsi di vari gradi di attività trascrizionale.,
I DSB possono stimolare l’HDR per consentire una sostituzione altamente precisa della regione danneggiata con un donatore omologo. Per ottenere la sostituzione mirata del DNA genomico, abbiamo introdotto una coppia di gRNAs, Cas9 e un donatore lineare con omologia alla regione mirata nelle cellule (Figura 4A). Il donatore lineare è stato ottenuto mediante amplificazione PCR con primer con una sequenza omologa di 50 bp. Questo stesso donatore è stato inserito con successo utilizzando un sistema di riparazione HDR basato su ZFN (18)., Per testare la fattibilità della sostituzione mediata da CRISPR / Cas9 con HDR, abbiamo mirato al locus CCL2 con una coppia di GRNA (#39 e #1854 mostrati in Figura 3) e un donatore con la sequenza di codifica EGFP (enhanced green fluorescent protein) e il sito SV40 poly(A) (Figura 4A; sequenze e posizioni sono presentate nel materiale supplementare). Usando questo sistema, circa lo 0,5% delle cellule mirate era EGFP-positivo, mentre solo lo 0,023% era EGFP-positivo nelle cellule di trasfezione simulate (appena trasfettate con donatore), che era simile alle cellule di controllo (0,021%, senza trasfezione)., Le cellule EGFP-positive sono state quindi ordinate per citometria a flusso. L’integrazione site-specific è stata confermata mediante PCR utilizzando due coppie di primer che fiancheggiano sia i bracci omologhi che l’intera regione sostituita. Come mostrato in Figura 4B, abbiamo osservato la regione sostituita prevista contenente la sequenza EGFP a tutta lunghezza e i bracci omologhi (risultato del sequenziamento Sanger mostrato nel materiale supplementare). È stato anche rilevato l’allele endogeno di tipo selvaggio (Figura 4B), indicando che non tutti gli alleli sono mirati., Inoltre, abbiamo selezionato singoli cloni dalle cellule EGFP-positive e abbiamo scoperto che tutti i cloni (6 su 6 esaminati) avevano l’integrazione prevista (Figura 4C), ma l’allele endogeno wild-type è stato rilevato anche in tre dei cloni (Figura 4C), suggerendo che solo un allele è stato preso di mira in quei cloni. L’espressione della proteina EGFP nelle cellule mirate (cellule ordinate EGFP-positive) è stata up-regolata durante il trattamento con TNF-α come valutato da Western blot e fluorescence activated cell sorting (FACS) (Figura 4, D ed E)., Questi risultati hanno dimostrato che il sistema CRISPR/Cas9 può essere utilizzato per creare sostituzioni gene/dominio con alta efficienza e precisione.

(A) Diagrammi schematici che descrivono la procedura di sostituzione genica mirata utilizzando CRISPR/Cas9 in cellule umane., Per testare l’efficacia della sostituzione genica mirata, gli RNA guida (GRNA) sono stati progettati per eliminare la regione indicata (contrassegnata dai siti 1 e 2) del gene CCL2 e per sostituire la regione eliminata con il donatore di cassette EGFP-polyA con braccia con brevi regioni di omologia. I siti mirati del sito 1 e del sito 2 sono #39 e #1854 all’interno del gene CCL2 mostrato in Figura 3. Le sequenze omologhe (50 bp) sono appena a monte ea valle dei siti di delezione. Le posizioni e le sequenze in dettaglio sono presentate nel Materiale supplementare., (B) L’efficienza della sostituzione genica mirata con CRISPR/Cas9 è stata determinata mediante PCR in cellule HEK 293T. I primer che coprono le giunzioni tra CCL2 ed EGFP sono stati utilizzati per l’amplificazione della PCR. C) Analisi PCR per la sostituzione genica mirata dei singoli cloni. (D,E) L’espressione della proteina EGFP su aggiunta di TNF-α nelle cellule HEK 293T è stata determinata da (D) Western blot e (E) FACS.
Qui descriviamo un approccio semplice ed efficiente per la delezione genica utilizzando il sistema CRISPR / Cas9., Abbiamo dimostrato che l’introduzione di questo sistema nelle cellule umane HEK 293T e in altri tipi di cellule umane ha indotto eliminazioni di frammenti fino a 10 kb con efficienze comprese tra l ‘ 11% e il 68%, a seconda della sequenza mirata. La capacità di eliminare in modo efficiente e preciso i segmenti genomici faciliterà lo studio degli elementi genomici funzionali nelle cellule umane. Questo approccio può essere potenzialmente utilizzato per colpire qualsiasi loci genomico.
C’è stata preoccupazione per la specificità del sistema CRISPR/Cas9 (19-21)., Per escludere fenotipi indesiderati dovuti a mutazioni off-target, suggeriamo di utilizzare almeno due diverse coppie di GRNA per ciascuna regione target. Nel nostro studio, la necessità di più coppie gRNA non era una limitazione importante, data la semplicità e l’elevata efficienza di questo sistema. È da notare che diverse coppie di GRNA destinate alla stessa regione hanno funzionato con elevata efficienza (figure 2 e 4). Un altro approccio per evitare mutazioni indesiderate è l’uso del metodo double nickase (22, 23)., Abbiamo anche applicato con successo il metodo double nickase per generare la delezione del DNA genomico, ma l’efficienza era notevolmente inferiore.
È noto che la riparazione del DNA DSBs è in gran parte mediata da NHEJ soggetto a errori, in cui le due estremità vengono elaborate e legate insieme in un modo che è spesso accompagnato da inserzioni e cancellazioni nucleotidiche. Tale giunzione finale soggetta a errori è stata osservata nella riparazione di DSB creati da ZFNs o TALENs. Al contrario, la riparazione dei DSB generati da Cas9 e due GRNA era molto precisa., I nostri risultati suggeriscono che le interruzioni sono direttamente legate senza elaborazione finale, rivelando un vantaggio precedentemente non apprezzato del percorso NHEJ. Resta da determinare il meccanismo che determina le legature precise. Una possibilità è che l’eliminazione mirata utilizzando Cas9 e due gRNAs si traduca in una giunzione che non viene riconosciuta da nessuno dei gRNAs originali. Abbiamo anche analizzato l’efficienza nella generazione di mutazioni indel per la singola coppia gRNA e gRNA (#39 e #224 gRNA in Figura 3A) mediante sequenziamento Sanger di ampliconi PCR (clonazione TA)., Di interesse, abbiamo osservato che l’efficienza di generare mutazioni indel per singolo gRNA era piuttosto bassa (9,5%, 2 di 21 cloni per #39 gRNA; 5%, 1 di 20 cloni per #224 gRNA). Tuttavia, la coppia gRNA ha generato un’elevata efficienza delle mutazioni indel (50%, 10 su 20 cloni per #39 e #224), che era simile al test utilizzando qPCR (52%, Figura 3B). Proponiamo che un singolo gRNA si traduca spesso in un’estremità smussata del sito di scissione, che verrà riparata con precisione da NHEJ. Pertanto, l’efficienza della generazione della mutazione è molto più bassa con l’uso di un singolo gRNA rispetto a una coppia di GRNA.,
Riconoscimenti
Questo lavoro è stato sostenuto da sovvenzioni del National Institute of Health (n. DP1CA174421) e la fondazione W. M. Keck a C.-Z. C, e National Natural Science Foundation of China (No. 81101481) e Shanghai Medical Talent Training Program (no. XYQ2011048) a S. L. H. Questo documento è soggetto alla Politica di accesso pubblico NIH.
Interessi concorrenti
Gli autori non dichiarano interessi concorrenti.
Dati supplementari
Per visualizzare i dati supplementari che accompagnano questo documento si prega di visitare il sito web della rivista all’indirizzo: www.,future-science.com/doi/suppl/10.2144/000114196