ヒト細胞におけるCRISPR/Cas9システムを用いた正確な遺伝子欠失と置換
ここでは、Cas9と組み合わせた二つのガイドRnaが効率的に欠失の修復が主に正確なエンド結合によって達成されるプロセスでヒト細胞における最大10kbのDNA欠失を生成することを示している。 さらに、我々はCRISPR/Cas9システムが線形相同修復ドナーの存在下で大きなゲノム断片を置き換えることができることを示すデータを提供します。,
クラスタ化された細菌は、定期的に短い回文繰り返し/CRISPR関連(CRISPR/Cas)遺伝子座は、ウイルスやプラスミド(1、2)の侵入から細胞を保護するRNA誘導免疫系をコード 化膿レンサ球菌では、II型CRISPR/Casシステムは、標的DNA配列の部位特異的切断を触媒するために、RNAガイドエンドヌクレアーゼ(RGEN)、Cas9を使用する。, Cas9の特定のゲノム部位へのターゲティングは、関連するCRISPR RNA(crRNA)内の20ヌクレオチドガイド配列によって媒介され、CrrnaをCas9複合体に募集するトランス活性化crRNA(tracrRNA)を必要とする(3)。 切断部位の認識は、crRNA-DNA塩基対形成およびdna相補領域に並置された三ヌクレオチド配列(NGG)であるプロトスペーサ隣接モチーフ(PAM)によって決定される(4)。, TracrRNA-crRNA複合体を模倣する単一ガイドRNA(gRNA)は、Cas9を標的とするゲノム部位にリクルートし、DNAにおける二本鎖切断(Dsb)を生成することができることは注目に値する(5)。 CRISPR/Cas9システムは、多様な細胞型および生物における部位特.なゲノム編集に適応されている(6-12)。
CRISPR/Cas9によるゲノム編集は、RNAプログラムされたRGENを用いて標的とされたゲノム遺伝子座にDSBを導入することによって開始される。 これに続いて、相同性指向修復(HDR)または非相同性末端結合(NHEJ)のいずれかを介してDSBの修復が続く。, 相同修復ドナーの存在下では、CRISPR/Cas9システムを使用して、HDRプロセスを通じて、標的遺伝子座における正確かつ定義された修飾および挿入を生成することができる。 相同修復ドナーが存在しない場合、CRISPR/Cas9によって生成された単一のDsbは、挿入または欠失(indel)突然変異をもたらすエラーが発生しやすいNHEJを介して修復 エクソンをコードするIndel変異は、早期停止コドンまたはフレームシフト変異を導入し、対応するタンパク質を不活性化する可能性がある。, 単一のDSBの修復から生成されたIndel変異は、タンパク質コード遺伝子の機能ドメインを特徴付けること、または遺伝子間またはイントロン調節配列または非コードRNA遺伝子などのゲノム要素の不活性化を目的とした実験には有用ではないかもしれない。 標的遺伝子座におけるDNA断片欠失は、これらの機能的要素を研究する手段を提供するであろう。 この目的のために、複数のDsbは、低効率ではあるが、ショウジョウバエ(12、13)、ゼブラフィッシュ(14)、およびヒト細胞(8)の欠失を生成するために導入されてい, 標的化されたゲノムDNA欠失はまた、ヒト細胞(15-17)における亜鉛フィンガーヌクレアーゼ(ZFN)または転写活性化剤様エフェクターヌクレアーゼ(TALEN)を用いて達成されている。 しかしながら、これらのアプローチの効率は一般に低い。 さらに、ZfnおよびTalenは、細胞の文脈において設計、開発、および経験的にテストするのがやや困難で高価なままである。
ここでは、CRISPR/Cas9システムによって触媒されるヒト細胞におけるフラグメント欠失の生成を調べた。 我々は、2grna Cas9と組み合わせて効率的に10kbまでのDNA欠失を作成することができますことを示しています。, 利益または修理のこの削除処理は単精度末の全衛生管理部”を新設しました。 さらに、CRISPR/Cas9による標的化された欠失は、標的遺伝子座の転写状態とは無関係であるように見える。 最後に、我々はCRISPR/Cas9システムは、線形相同修復ドナーの存在下で大きなゲノム断片を置き換えるために使用することができることを示しています。
材料および方法
プラスミド構築
基本的なH1プロモーターをpLVTHMプラスミド(Addgene,#12247,Cambridge,MA)から増幅した。, 修飾されたH1プロモーターと二つのBsaIサイトを持つ所望のgRNA配列のバックボーンを含むオリゴヌクレオチドを合成した(PAN Facility、スタンフォード大学)。 得られた全長生成物をPCRによって増幅し、pUC19ベクターにクローニングした。 アンピシリン遺伝子(amp)とPuc19ベクターのH1プロモーターはBsaI制限酵素部位を含み、これらは変異した(amp遺伝子はアミノ酸配列を変更しないG1601Cから変更され、H1プロモーターはGAGACCからGAGGACCに変更された)BsaI部位を排除した。, Grnaクローニングのためのプロトコールを補足資料に示した。 すべてのターゲティングサイト配列を補足表S1に示す。
細胞培養
HEK293T、SK-Hep1、およびHeLa細胞を、10%ウシ胎児血清(FBS)(Hyclone、Logan、UT)およびペニシリン/ストレプトマイシン(pen/strep)(Invitrogen、Carlsbad、CA)を補充したDulbecco”s modified Eagle”s培地(DMEM)中で培養した。 PC3細胞をRPMI-1640培地中で10%FBSおよびpen/strepを添加して培養した。, 腫瘍壊死因子α(TNF-α)刺激のために、293t細胞を、示された濃度のTNF-α(R&D Systems、Minneapolis、MN)で処理した。 細胞は、加湿インキュベーター内で37℃および5%CO2に維持した。
標的とされたDNA欠失
HEK293T細胞は、12ウェルプレートにウェル当たり100,000細胞の密度で播種した。 24時間後、細胞に1μg Cas9プラスミド(Addgene、#41815)、0.5μg gRNA T1、および0.5μg gRNA T2プラスミドを、製造業者のプロトコルに従ってLipofectamine2000(Invitrogen)を用いて一時的にトランスフェクトした。, QuickExtract DNA抽出液(Epicentre Biotechnologies、Madison、WI)を使用して、トランスフェクション後48時間でゲノムDNAを抽出した。 標的領域に隣接するプライマーを使用して標的領域を増幅するために共通PCRを実施した。 野生型および切り捨てられたゲノム断片をゲル電気泳動により分解した。 リアルタイムPCR(RT-PCR)を行って、接合部を横切るか、または欠失領域内のプライマーを使用して欠失の割合を定量した。 比較Cq法を用いて、基準領域(ACTB遺伝子座)に対する標的領域の発現レベルを計算した。, 標的領域における欠失の割合を、対照細胞に対する標的細胞の比によってさらに計算した。 全てのプライマー配列を補足表S2に記載する。
ターゲットシーケンシング
細胞は、トランスフェクションの二日後に採取し、ゲノムDNAはQuickExtract DNA抽出溶液(Epicentre Biotechnologies)を用いて抽出した。 細胞由来のゲノムDNAで標的領域を増幅するためにPCRを実施し、増幅子をMiseq Personal Sequencer(Illumina,San Diego,CA)によって深く配列決定した。,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., ドナー配列は補足資料に示されている。 二つの5′-最もリンケージはホスホロチオ酸(アスタリスクで示される)である。 6ウェルプレート中の細胞は一時的に2.0μg Cas9プラスミド、0.8μg gRNA T1プラスミド、0.8μg gRNA T2プラスミド、および0.4μg線形ドナーリポフェクタミン2000(Invitrogen)を用いてトランスフェクトされた。 トランスフェクション後48時間で、細胞を1ng/mL TNF-αで24時間処理し、次いでGFP陽性細胞を選別した。
ルシフェラーゼアッセイ
ルシフェラーゼアッセイのために、HEK293T細胞は96ウェルプレートにウェルあたり5000細胞の密度で播種した。, 24時間後、細胞は一時的に5ngのpRL-TKレニラルシフェラーゼレポーターと100ngのルシフェラーゼレポーターサイトメガロウイルス(CMV)、SV40(シミアンウイルス40)、または 48時間後、ルシフェラーゼ活性は、デュアルルシフェラーゼレポーターアッセイシステム(Promega)で測定した。
ウェスタンブロット
タンパク質は、ドデシル硫酸ナトリウム—PAGE(SDS-PAGE)によって分離され、ニトロセルロース膜に転送されました。 膜を5%非脂肪乳でブロックし、GFP抗体(CST、#2555S、Danvers、MA)でインキュベートした。, 抗原-抗体複合体は増強された化学ルミネセンス試薬で検出された。
結果と議論
我々は、ヒト細胞におけるゲノムDNAを突然変異誘発する細菌II型CRISPR/Cas9システムを適応させました。 S.pyogenes Cas9タンパク質c末端SV40核局在シグナルを有するヒトコドン最適化バージョンは、以前に記載されたシステム(6)を用いて発現した。 所望の配列にCas9切断を指示するために、我々は、以下の変更されたヒトH1ポリメラーゼIIIプロモーターから、ガイドRna(gRNAs)と呼ばれるcrRNA-tracrRNA融合転写産物を発現した。, H1プロモーターの3’末端は、任意のヌクレオチドで始まるgrnaの転写を可能にするように変更された。 20bp crRNA標的の後にPAM配列NGG(ここで、Nは任意のヌクレオチド)が続くという要件によってのみ制約され、このアプローチは原則として、N20NGGという形を有する任意のゲノム位置を標的とするために使用することができる。 Grna発現ベクターのクローニングを容易にするために,Iis型制限酵素Bsaiを用いた。 これは、DNA上の標的部位に相補性の領域を含む24bpのオリゴヌクレオチドの合成を必要とした。, GRNA発現ベクターのクローニングのための簡単で効率的なプロトコル(図1A)は、補足資料に詳細に記載されています。
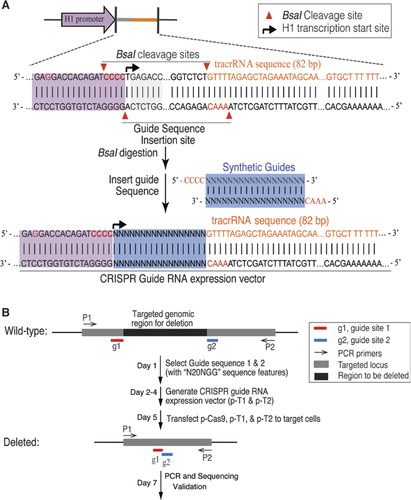
ゲノムDNAの大きなセグメントの欠失のために、我々は標的遺伝子座(図1B)に対するgrnaのペアを使用しました。 パターンN20NGGを持つ二つのターゲットサイトは、ターゲット領域の境界で選択されました。 Grna対の様々な組み合わせによって導かれた標的欠失の効率は、標的領域に隣接するプライマーを用いたPCR分析によって決定された。 野生型および切り捨てられたゲノム断片をゲル電気泳動によって分解した。 PCR増幅バイアスを回避するために、一つのプライマー対を用いてRT-PCRにより欠失率を定量した。, プライマーは、欠失接合部(欠失領域外の一つのプライマー、欠失領域内の他のプライマー)または欠失領域内(両方のプライマーが欠失領域内に位置する)を横切っ したがって、標的細胞および対照細胞の両方について、単一のバンドのみがプライマー対で増幅される。 本発明者らは、同じプライマー対によって増幅されたPCR産物(標的細胞と対照細胞)の相対量を比較することにより、欠失率を計算した。 標的欠失は、配列決定によってさらに検証された。,
gRNAペアがその後の修復および欠失の生成にどのように影響するかを評価するために、我々はまず、ヒトCDC42ゲノム遺伝子座を標的とし、約200から10,000bpまでの距離によって分離されたgrnaのセットを設計した(図2Aおよび補足表S1)。 次に、ヒトHEK293T細胞においてCas9の存在下で欠失を生成する各gRNA対の能力を評価した。 NHEJベースの欠失の堅牢な効率(最大68%)は、qPCRによって確認されました(図2B-2D)。, でも、10kbのゲノム領域の削除のために、我々はgRNAペアに応じて、16%から28%のターゲット率を得ました。 このRNAを介した編集プロセスは急速であり、最初に検出可能な欠失はトランスフェクション後約12時間に現れた(補足図S1)。 このシステムは、PC3、SK-Hep1、およびHeLa細胞を含む様々な細胞型において有効であった(補足図S2)。

(A)CDC42遺伝子座を標的とするガイドRna(grna)の位置を描いた模式図。 (B–D)CRISPR/Cas9による標的欠失の効率は、HEK293T細胞におけるPCRによって決定された。 予想される欠失領域外のプライマーを使用した。 欠失の割合は、接合部を横切るか、または欠失領域内のプライマーを使用してRT-PCRによって定量した。 (E–F)CDC42における標的削除の効率と精度は、(E)サンガーシーケンシングと(F)ハイスループットシーケンシング分析によって確認されました。 欠失アンプリコンのみを含むPCR産物を配列決定のために濃縮した。,
欠失は、予想される切断部位にまたがるPCR産物を配列決定することによってさらに確認された。 Sangerシーケンシングは、Cas9によって作成された鈍端Dsbの正確な結紮に起因する欠失接合を示した;各DSBは正確に3bp pamシーケンスの上流に発生した(図2Eおよび補足図S3)。 また、欠失アンプリコンのディープシーケンシングを用いて欠失効率の精度を評価し、読み取りの約80%において、目標とされたDsbは完全に修復された(図2F)。,
我々は、hek21T細胞におけるmicroRNA miR-293遺伝子を含むゲノム遺伝子座から断片を削除するように設計されたgRNAペアをテストすることにより、これらの知見 二つのgrnaは、miR-21ヘアピン(補足図S4)の境界をターゲットに設計されました。 欠失効率は、PCRアッセイを用いて測定したところ、二つのgrnaおよびCas9とのトランスフェクション後の38%であった(補足図S4B)。 深い配列の確認、削除が正確に期待した(補正図S4C).,
CRISPR/Cas9媒介遺伝子欠失システムが標的遺伝子の転写状態によって影響されるかどうかを調べるために、ケモカイン(C-Cモチーフ)リガンド2(CCL2)をコードす CCL2はCCケモカインファミリーに属する小さなサイトカインであり、CCL2遺伝子はNF-κbシグナル伝達の標的である。 我々は、CCL2遺伝子座の5’および3’末端に位置する八つの標的部位をランダムに選択した(図3A)。 我々は、Cas9およびHEK293T細胞におけるgrnaの異なるペアを用いて、遺伝子の異なる領域の堅牢かつ効率的な欠失を達成した(図3B–3D)。,

(A)CCL2遺伝子座を標的とするガイドRna(grna)の位置を描いた模式図。 (B–D)CCL2のCRISPR/Cas9によるHEK293T細胞における標的欠失の効率。 (E)CCL2mRNAレベルは、HEK293T細胞における定量的RT-PCR分析によって腫瘍壊死因子α(TNF-α)添加時に決定された。 データを平均±semで三重に示した。, (F)CRISPR/Cas9によるCCL2の標的欠失の効率は、HEK24時間に対するTNF-αによる処理後の293T細胞における。 (G)サイトメガロウイルス(CMV)、またはSV40、またはHEK293T細胞における塩基性プロモーターからのルシフェラーゼ活性のレベル。 データを平均±semで三重に示した。 (H)CMV、またはSV40、またはHEK293T細胞における塩基性プロモーターによって制御されるルシフェラーゼ遺伝子の標的欠失の効率。 (B–D、F、H)欠失の割合を、接合部を横切るか、または欠失領域内のプライマーを使用してRT-PCRによって定量した。,
CCL2遺伝子の発現は劇的にTNF-α(最大≥300倍の増加)HEK293T細胞(図3E)によって誘導され、CRISPR/Cas9を介した標的ゲノム編集が転写活性によって影響されるかどうかを調べるための良いモデルを提供する。 興味深いことに、CCL2遺伝子座の標的欠失の効率は、TNF-αによる細胞の処理によって影響されなかった(図3F)、転写がCRISPR/Cas9媒介欠失を変化させなかったことを示唆している。, さらにこの結果を確認するために、我々は異なる転写活性がルシフェラーゼアッセイ(図3G)を用いて評価することができる異なる強さを持つ様々なプロモーターによって駆動される外因性レポーター遺伝子を標的とした。 PCRアッセイは、レポーター遺伝子をCas9およびgRNAペアと共にトランスフェクションした後、HEK293T細胞における標的欠失において同様の効率を明らかにした(図3H)。 この結果は、転写活性の様々な程度の発生にもかかわらず、NHEJ媒介修復が起こり得ることを示している。,Dsbは、HDRを刺激して、損傷領域を相同ドナーによる高精度な置換を可能にすることができる。 標的化されたゲノムDNA置換を得るために、我々は、対のgrna、Cas9、および標的化された領域に相同性を有する線形ドナーを細胞に導入した(図4A)。 線形ドナーは、50bpの相同配列を有するプライマーを用いたPCR増幅によって得られた。 この同じドナーは、ZFNベースのHDR修復システム(18)を使用して正常に挿入されました。, HDRによるCRISPR/Cas9媒介置換の実現可能性をテストするために、我々はccl2遺伝子座grnaのペア(#39と#1854図3に示す)と強化された緑色蛍光タンパク質(EGFP)コーディングシーケンスとSV40ポリ(a)サイトを有するドナーを標的とした(図4A;配列と位置は補足材料に提示されている)。 このシステムを使用して、標的細胞の約0.5%がEGFP陽性であったが、0.023%のみが対照細胞(0.021%、トランスフェクションなし)と同様であった模擬トランスフェクション細胞(ドナーでトランスフェクションされた)においてEGFP陽性であった。, 次いで、EGFP陽性細胞をフローサイトメトリーによって選別した。 相同腕と置換領域全体の両方に隣接する二対のプライマーを用いたPCRにより部位特異的統合を確認した。 図4Bに示すように、我々は、全長EGFP配列および相同アームを含む予想される置換領域を観察した(補足材料に示されるSanger配列決定の結果)。 内因性の野生型対立遺伝子も検出され(図4B)、すべての対立遺伝子が標的とされているわけではないことを示している。, さらに、EGFP陽性細胞から単一のクローンを選択し、すべてのクローン(6の6を調べた)が期待される統合を持っていたことがわかった(図4C)が、内因性野生型対立遺伝子も三つのクローン(図4C)で検出され、それらのクローンでは一つの対立遺伝子のみが標的とされたことを示唆している。 標的細胞(EGFP陽性選別細胞)におけるEGFPタンパク質の発現は、ウェスタンブロットおよび蛍光活性化細胞選別(FACS)によって評価されるようにTNF-α処理でアップレギュレートされた(図4、DおよびE)。, これらの結果は、CRISPR/Cas9システムを使用して、高い効率と精度で遺伝子/ドメイン置換を作成できることを示しました。

(A)ヒト細胞におけるCRISPR/Cas9を用いた標的遺伝子置換の手順を描いた模式図。, 標的遺伝子置換の有効性をテストするために、ガイドRna(grna)は、ccl2遺伝子の示された領域(サイト1および2でマークされた)を削除し、相同性の短い領 サイト1およびサイト2の標的部位は、#39および#1854であり、図3に示すCCL2遺伝子内である。 相同配列(50bp)は、欠失部位のちょうど上流および下流である。 詳細な位置および順序は、補足資料に示されている。, (B)CRISPR/Cas9による標的遺伝子置換の効率を、HEK293T細胞におけるPCRによって決定した。 CCL2とEGFPの間の接合部にまたがるプライマーは、PCR増幅のために使用されました。 (C)単一クローンの標的遺伝子置換のためのPCRアッセイ。 (D,E)HEK293T細胞におけるTNF-α添加によるEGFPタンパク質の発現を、(D)ウェスタンブロットおよび(E)FACSによって決定した。
ここでは、CRISPR/Cas9システムを使用して遺伝子欠失のためのシンプルで効率的なアプローチについて説明します。, 我々は、ヒトHEK293T細胞、および他のヒト細胞型へのこのシステムの導入は、目標とされた配列に応じて、10%と68%の間の範囲の効率で最大キロバイトの断片の欠失を誘導することを実証した。 効率的かつ正確にゲノムセグメントを削除する能力は、ヒト細胞における機能的なゲノム要素の研究を容易にするでしょう。 このアプローチを採ることが潜在的に使用を対象にしたゲノム遺伝子座.
CRISPR/Cas9システム(19-21)の特異性に関する懸念があった。, 標的外突然変異による望ましくない表現型を除外するために、少なくとも二つの異なる対のgrnaを各標的領域に使用することを示唆した。 我々の研究では、複数のgRNA対の必要性は、このシステムのシンプルさと高効率を考えると、大きな制限ではなかった。 同じ領域をターゲットにした異なる対のgrnaが高効率で働いていたことは注目に値する(図2および図4)。 望ましくない突然変異を避けるための別のアプローチは、ダブルニッカーゼ法(の使用である22、23)。, また,ダブルニッカーゼ法を適用してゲノムDNAの欠失を生成することに成功したが,効率はかなり低かった。DNA Dsbの修復は、エラーを起こしやすいNHEJによって主に媒介されることが知られており、このNHEJでは、両端がヌクレオチドの挿入および欠失を頻繁に伴う方法で処理され、一緒に連結される。 このようなエラーが発生しやすい端部結合は、ZfnまたはTalenによって作成されたDsbの修復において観察された。 対照的に、Cas9と二つのgrnaによって生成されたDsbの修復は非常に正確でした。, 我々の結果は、ブレークが直接NHEJ経路の以前に評価されていない利点を明らかに、エンドプロセシングなしで結紮されていることを示唆している。 正確な結紮をもたらすメカニズムは依然として決定されている。 一つの可能性は、Cas9と二つのgrnaを使用して標的削除は、元のgrnaのいずれかによって認識されないジャンクションになるということです。 また、PCRアンプリコンのSangerシーケンシング(TAクローニング)により、個々のgRNAおよびgRNAペア(#39および#224gRNA図3A)のindel変異を生成する効率を分析しました。, 興味深いことに、我々は、単一gRNAのインデル変異を生成する効率が非常に低かったことを観察した(9.5%、2の21クローン#39gRNA;5%、1の20クローン#224gRNA)。 しかしながら、gRNA対は、qpcrを用いたアッセイと同様であったインデル変異(50%、10の20クローン#39および#224)の高効率を生成した(52%、図3B)。 我々は、単一のgRNAは、多くの場合、NHEJによって正確に修復される切断部位の一方の鈍い端をもたらすことを提案します。 したがって、突然変異を生じる効率は、一対のgrnaよりも単一のgrnaの使用によってはるかに低い。,
謝辞
この作品は、国立衛生研究所からの助成金によってサポートされています(No。 DP1CA174421)およびC.-Z.CへのW.M.Keckの基礎、および中国(第81101481)および上海の医学の才能のトレーニングプログラム(いいえ。 XYQ2011048)to S.L.H.このペーパーはNIHパブリックアクセスポリシーの対象となります。
競合する利益
著者は競合する利益を宣言していません。
補足データ
この論文に付随する補足データを表示するには、ジャーナルのウェブサイトwwwをご覧ください。,future-science.com/doi/suppl/10.2144/000114196