정확한 유전자를 삭제 및 교체를 사용하는 CRISPR/Cas9 시스템 인간의 세포에서
여기서 우리는 두 가이드 RNAs 와 결합 Cas9 효율적으로 생성하 DNA 삭제까지 10kb 에서 인간의 세포에서 프로세스는 복구의 삭제를 주로 수행하여 정확한 끝입니다. 또한,우리는 데이터를 제공하는 CRISPR/Cas9 시스템을 대체할 수 있습 큰 게놈 조각에서 존재의 선형 상동 복구 기증합니다.,
세균 클러스터 정기적으로 interspaced 짧은 회문을 반복/CRISPR 관련(CRISPR/Cas)loci 인코딩 RNA 유도 면역체계를 보호하는 세포를 침입에 대하여 바이러스 및 플라스미드(1,2). Streptococcus pyogenes 에서 type II CRISPR/Cas 시스템은 표적 DNA 서열의 부위 특이 적 분열을 촉매하기 위해 RNA-guided endonuclease(RGEN),Cas9 를 사용합니다., 의 대상으로 Cas9 특정 사이트는 게놈은 중재에 의해 20 뉴클레오티드 가이드는 순서에 관련된 CRISPR RNA(crRNA)을 필요로하는 트랜스를 활성화하 crRNA(tracrRNA)는 신참 crRNA 로 Cas9 복잡한(3). 인식 분열의 사이트에 의해 결정됩 crRNA-DNA 기반의 페어링 및 protospacer 초고속 무선 인터넷을 무료로 이용 모티브(PAM),세 개의 염기서열(NGG)juxtaposed DNA 보완적인 지역(4)., 그것은 주목할 만하는 단일 guide RNA(gRNA)을 모방하는 tracrRNA-crRNA 복잡할 수 있습을 모집 Cas9 을 대상으로 게놈 사이트를 생성 더블 좌초 breaks(DSBs)에서 DNA(5). CRISPR/Cas9 시스템은 다양한 세포 유형 및 유기체(6-12)에서 사이트 특이 적 게놈 편집에 적합했습니다.
Crispr/Cas9 를 이용한 게놈 편집은 RNA-프로그래밍 된 RGEN 을 사용하여 표적화 된 게놈 로커에서 DSB 의 도입으로 시작됩니다. 그 다음에는 HDR(homology-directed repair)또는 NHEJ(nonhomologous end-joining)를 통해 DSB 를 복구합니다., 의 존재에서 동종 수리,기증자는 CRISPR/Cas9 시스템을 사용할 수 있습을 생성하고 정확한 정의을 수정하고 삽입에서는 대상 소재를 통해 HDR 과정입니다. 의 부재에서 동종 수리,기증자 단 DSBs 에 의해 생성된 CRISPR/Cas9 는 복구를 통해 오류가 발생하기 쉬운 NHEJ 는 결과를 삭제하거나 삽입(indel)돌연변이가 있습니다. Indel 돌연변이 코딩에서 엑손할 수 있습을 소개하는 조지 코돈나 프레임-이동 돌연변이,함으로써 비활성화 해당하는 단백질이다., Indel 돌연변이 생성되는 수리에서 단일 DSB 되지 않을 수 있습에서 유용한 실험을 목표로 특성화 기능 도메인 단백질의 코딩하는 유전자를 위해 또는 비활성화의 genomic 등의 요소 intergenic 또는 intronic 규제 시퀀스 또는 noncoding RNA 유전자입니다. 표적 loci 에서의 DNA 단편 삭제는 이러한 기능적 요소를 연구 할 수있는 길을 제공 할 것이다. 이를 위해,여러 DSBs 도입되었을 생성하는 삭제에서는 초파리(12,13),zebrafish(14),그리고 인간 세포(8),이기는 하지만 저렴한 효율입니다., 대상 genomic DNA 삭제되었을 사용하여 달성 아연락 nuclease(ZFN)또는 전송 활성화 같은 이펙터 nuclease(탈렌)인간의 세포에서(15-17). 그러나 이러한 접근법의 효율성은 일반적으로 낮습니다. 또한,ZFNs 및 TALENs 는 세포 맥락에서 설계,개발 및 경험적으로 테스트하기가 다소 어렵고 비용이 많이 든다.
여기에서,우리는 CRISPR/Cas9 시스템에 의해 촉매 된 인간 세포에서 단편 삭제의 생성을 조사했다. 우리는 Cas9 와 결합 된 2 개의 gRNAs 가 최대 10kb 의 DNA 삭제를 효율적으로 만들 수 있음을 보여줍니다., 관심의,우리는이 삭제 프로세스의 복구는 주로 정확한 최종 결합에 의해 수행되는 것을 발견했다. 더욱이,CRISPR/Cas9 로 표적화 된 삭제는 표적화 된 궤적의 전사 상태와는 독립적 인 것으로 보인다. 마지막으로,우리는 CRISPR/Cas9 시스템을 대체하기 위해 사용될 수 있습 큰 게놈 조각에서 존재의 선형 상동 복구 기증합니다.
재료 및 방법
플라스미드 건설
기본 H1 발기인에서 증폭 pLVTHM 플라스미드(Addgene,#12247,Cambridge,MA)., 2 개의 BsaI 부위를 갖는 원하는 gRNA 서열의 변형 된 H1 프로모터 및 백본을 함유하는 올리고 뉴클레오티드가 합성되었다(Pan Facility,Stanford University). 생성 된 전장 생성물을 PCR 에 의해 증폭시키고 pUC19 벡터로 클로닝 하였다. 의 암피실린 유전자(a)및 H1 발기인에 pUC19 벡터 포함 BsaI 제한효소 사이트 이들은 돌연변이(amp 유전자에서 변경 G1601C 하지 않는 아미노산 시퀀스를 변경;H1 발기인이 변경되었에서 GAGACC 을 GAGGACC)을 제거하 BsaI 사이트입니다., GRNA 복제에 대한 프로토콜은 보충 자료에 제시되어있다. 모든 타겟팅 사이트 시퀀스는 보충 표 S1 에 제시되어있다.
셀 문화
HEK293T,SK-Hep1,과 헬라 세포 배양에 Dulbecco”s 정의 독수리”s 매체(DMEM)으로 보충된 10%에 태아 소럼(FBS)(Hyclone,Logan,UT)과 페니실린/수트렙토마이신(펜/염)(Invitrogen,Carlsbad,CA). PC3 세포를 10%FBS 및 pen/strep 로 보충 된 RPMI-1640 배지에서 배양 하였다., 종양 괴사 인자 α(TNF-α)자극,293T 세포 치료를 했으로 표시된 농도의 TNF-α(R&D 시스템,Minneapolis,MN). 세포를 가습 배양기에서 37°c 및 5%CO2 로 유지 하였다.
표적화 된 DNA 삭제
hek293T 세포를 웰 당 100,000 세포의 밀도로 12-웰 플레이트에서 시드 하였다. 후 24h,셀 transiently 페 1µg Cas9 플라스미드(Addgene,#41815),0.5μg gRNA T1,0.5μg gRNA T2 플라스미드를 사용하여 Lipofectamine2000(Invitrogen)as per the manufacturer”s 프로토콜., 게놈 DNA 는 QuickExtract DNA 추출 용액(Epicentre Biotechnologies,Madison,WI)을 사용하여 transfection 후 48h 를 추출 하였다. 일반적인 PCR 은 표적화 된 영역을 측면으로하는 프라이머를 사용하여 표적화 된 영역을 증폭시키기 위해 수행되었다. 야생형 및 잘린 게놈 단편은 겔 전기 영동에 의해 해결되었다. 실시간 PCR(RT-PCR)를 수행 되었을 정량화하의 퍼센트는 프라이머를 사용하여 삭제에서 교차점 이내에 또는 삭제 지역입니다. 비교 Cq 방법은 기준 영역(ACTB locus)에 상대적인 대상 영역의 발현 수준을 계산하는 데 사용되었습니다., 표적 영역에서의 삭제의 퍼센트는 대조군 세포에 상대적인 표적 세포의 비율에 의해 추가로 계산되었다. 모든 프라이머 서열은 보충 표 S2 에 나열되어있다.
목표 시퀀싱
세포 수확틀 transfection 후,그리고 genomic DNA 를 사용하여 추출 QuickExtract DNA 를 추출 솔루션(중심지로 바이오). PCR 은 세포에서 유래 된 게놈 DNA 로 표적화 영역을 증폭시키기 위해 실시되었으며,amplicons 는 MiSeq Personal Sequencer(Illumina,San Diego,CA)에 의해 심층 시퀀싱되었다.,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., 기증자 서열은 보충 자료에 제시되어있다. 두 개의 5′-대부분의 연결 고리는 포스 포로 티오 에이트(별표로 표시)입니다. 6-웰 플레이트의 세포를 lipofectamine2000(Invitrogen)을 사용하여 2.0μg Cas9 플라스미드,0.8μg gRNA T1 플라스미드,0.8μg gRNA T2 플라스미드 및 0.4μg 선형 공여체로 과도하게 형질 전환시켰다. Transfection 후 48h 에서 24h 에 대해 1ng/mL TNF-α 로 세포를 처리 한 다음 GFP 양성 세포를 분류 하였다.
루시페라아제 분석
에 대한 루시페라아제 분석 결과,HEK293T 세포에서 시드 96-well 플레이트에서 밀도의 5000 당 셀습니다., 후 24h,셀 transiently transfected5ng 의 pRL-TK Renilla 루시페라아제 기자 100ng 의 루시페라아제 기자와 세포 확대 바이러스(CMV),SV40(Simian virus40),또는 기본 발기인이다. 48 시간 후,이중 루시퍼 라제 리포터 분석 시스템(Promega)으로 루시퍼 라제 활성을 측정 하였다.
웨스턴 오
단백질 분리하여 나트륨 라우릴 설페이지(SDS-PAGE)전송하는 니트로 막. 막을 5%무 지방 우유로 차단하고 GFP 항체(CST,#2555S,Danvers,MA)로 배양 하였다., 항원-항체 복합체는 강화 된 화학 발광 시약으로 검출되었다.
결과 및 토론
우리가 적응 세균 type II CRISPR/Cas9 시스템 mutagenize genomic DNA 인간의 세포에서. C 말단 SV40 핵 국소화 신호를 갖는 S.pyogenes Cas9 단백질의 인간 코돈 최적화 버전은 이전에 기술 된 시스템(6)을 사용하여 발현되었다. 직 Cas9 열을 원하는 순서,우리는 표현 crRNA-tracrRNA 퓨전 성적증명서,이하라고 가이드 RNAs(gRNAs)에서,수정의 인간 H1polymerase III 발기인이다., H1 프로모터의 3’말단은 임의의 뉴클레오타이드로 시작하는 gRNAs 의 전사를 허용하도록 변형되었다. 제한에 의해서만 요구하는 20bp crRNA 대상에 따라야 PAM 시퀀스,NGG(N 은 어떤 뉴클레오티드)이 방식에서 할 수 있는 원칙을 대상으로 사용될 모든 게놈 위치에 있는 양식을 N20NGG. GRNA 발현 벡터의 복제를 용이하게하기 위해,우리는 유형 IIs 제한 효소 인 BsaI 를 사용했다. 이것은 DNA 상의 표적 부위에 상보성의 영역을 포함하는 24bp 올리고 뉴클레오타이드의 합성을 필요로 하였다., GRNA 발현 벡터(그림 1A)의 복제에 대한 간단하고 효율적인 프로토콜은 보충 자료에 자세히 설명되어 있습니다.
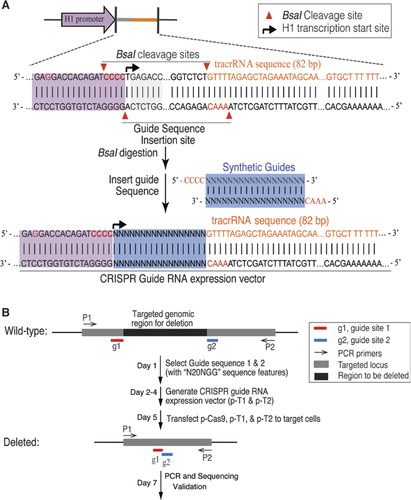
(A)가이드 RNA(gRNA)발현 벡터의 설계. 벡터는 트랜스 활성화 RNA/CRISPR RNA(tracrRNA)에 융합 된 합성 gRNA 를 갖는 gRNA 성적표를 생성하도록 설계되었다., H1 프로모터는 h1 프로모터에서 GAGACC 를 GAGGACC 로 변경함으로써 내부 유형 IIS 제한 효소 BsaI 부위를 제거하도록 변형되었다. A BsaI 사이트 소개되었을 만드는 복제에 대한 사이트 gRNA 및 tracrRNA fusions 의 삽입에 의해 합성 oligonucleotide duplexes 호환 있습니다. H1 프로모터의 3’말단은 5’말단에서 임의의 뉴클레오타이드로 gRNAs 의 전사를 용이하게하기 위해 CCACAGATCCCC 로 변형되었다. (B)CRISPR/Cas9 로 표적화 된 유전자 결실의 단계.,
게놈 DNA 의 큰 세그먼트의 삭제를 위해,우리는 표적화 된 locus 에 대해 한 쌍의 gRNAs 를 사용했다(그림 1B). 패턴 N20NGG 를 갖는 2 개의 타겟 사이트가 타겟 영역의 경계에서 선택되었다. 효율성의 대상으로 삭제 가이드에 의해 다양한 조합의 gRNA 쌍들에 의해 결정 PCR 분석을 사용하여 프라이머 측면을 대상으로 영역입니다. 야생형 및 잘린 게놈 단편은 겔 전기 영동에 의해 해결되었다. PCR 증폭 바이어스를 피하기 위해,percent deletion 은 하나의 프라이머 쌍을 사용하여 RT-PCR 에 의해 정량화되었다., 프라이머 설계에서 삭제 접합(하나의 프라이머 외부 삭제 영역이고,다른 프라이머를 이내에 삭제 지역)또는 삭제 영역(모두 프라이머를 찾아에서 삭제 지역). 따라서,단일 밴드 만이 표적 세포 및 대조군 세포 모두에 대해 프라이머 쌍으로 증폭된다. 우리는 계산%를 삭제를 비교하여 상대적 양의 PCR 제품(목표 세포와 제어 세포)에 의해 증폭되는 같은 프라이머다. 표적화 된 삭제는 시퀀싱에 의해 추가로 확인되었다.,
을 평가하는 방법 gRNA 쌍에 영향을 미칠 수 있 후속 복구와 세대의 삭제,우리는 먼저 설계 세트의 gRNAs 대상에 대한 인 CDC42genomic 궤적 분리에 의 거리에 이르기까지 약 200 10,000bp(그림 2A 및 보조 표 S1). 그런 다음 우리는 Cas9 의 존재 하에서 인간 HEK293T 세포에서 삭제를 생성하는 각 gRNA 쌍의 능력을 평가했다. NHEJ 기반 삭제의 강력한 효율성(최대 68%)은 qPCR 에 의해 확인되었습니다(그림 2B–2D)., 10kb 게놈 영역의 삭제에 대해서도,우리는 gRNA 쌍에 따라 16%에서 28%의 표적화 비율을 얻었다. 이 RNA 매개 편집 과정은 첫 번째 검출 가능한 삭제가 약 12 시간 후 transfection(보충 그림 S1)으로 나타나 급속했다. 이 시스템은 PC3,SK-Hep1 및 HeLa 세포(보충 그림 S2)를 포함한 다양한 세포 유형에서 효과적이었다.

(A)CDC42 궤적을 대상으로하는 가이드 RNAs(gRNAs)의 위치를 묘사하는 개략도. (B-D)CRISPR/Cas9 로 표적화 된 삭제 효율은 HEK293T 세포에서 PCR 에 의해 결정되었다. 예상 삭제 영역 외부의 프라이머가 사용되었습니다. 삭제의 퍼센트는 접합부를 가로 질러 또는 삭제 영역 내에서 프라이머를 사용하여 RT-PCR 에 의해 정량화되었다. (E–F)CDC42 에서의 표적 삭제의 효율 및 정밀도는(E)Sanger 시퀀싱 및(F)고 처리량 시퀀싱 분석에 의해 확인되었다. 삭제 앰플리콘만을 함유하는 PCR 생성물은 시퀀싱을 위해 농축되었다.,
삭제가에 의해 확인 시퀀싱 PCR 제품에 걸친 예상되는 협곡 사이트입니다. 생어 시퀀싱 보여 삭제 접합에서 결과의 정확한 결찰의 무뚝뚝을 종료 DSBs 에 의해 만들어 Cas9;각 DSB 발생했을 정확히 3bp 상류의 PAM 시퀀스(그림 2E 및 보조 도 S3)입니다. 우리는 또한 사용 깊은 시퀀싱 삭제 amplicons 의의 정확도를 평가 삭제 효율성에 대한의 80%를 읽고,대상 DSBs 었다 완벽한 수리는(그림 2).,
우리는 HEK293T 세포에서 microRNA miR-21 유전자를 포함하는 게놈 로커에서 단편을 삭제하도록 고안된 gRNA 쌍을 시험함으로써 이러한 발견을 재검토했다. 두 개의 gRNAs 는 miR-21 머리핀의 경계를 대상으로하도록 설계되었습니다(보충 그림 S4). 삭제 효율은 pcr 분석법(보충 그림 S4B)을 사용하여 측정 한 바와 같이 두 개의 gRNAs 와 Cas9 로 transfection 한 후 38%였다. 딥 시퀀싱은 삭제가 예상대로 정확하게 발생했음을 확인했습니다(보충 그림 S4C).,
여부를 조사하 CRISPR/Cas9 중재된 유전자를 삭제 시스템에 의해 영향을 받 transcriptional 상태의 타겟 유전자의 유전자로 인코딩하는 chemokine(C-C motif)ligand2(CCL2)를 대상으로 합니다. CCL2 는 CC 케모카인 계열에 속하는 작은 사이토 카인이다;CCL2 유전자는 NF-kB 신호 전달의 표적이다. 우리는 ccl2 유전자 locus 의 5’및 3’말단에 위치한 8 개의 표적 부위를 무작위로 선택했습니다(그림 3A). 우리는 HEK293T 세포에서 Cas9 와 다른 쌍의 gRNAs 를 사용하여 유전자의 다른 영역의 강력하고 효율적인 삭제를 달성했습니다(그림 3B–3D).,

(A)CCL2 궤적을 대상으로하는 가이드 RNAs(gRNAs)의 위치를 묘사하는 개략도. (B-D)HEK293T 세포에서 CRISPR/Cas9 로 CCL2 의 표적 삭제 효율. (E)CCL2mRNA 수준은 HEK293T 세포에서 정량적 RT-PCR 분석에 의해 종양 괴사 인자 α(TNF-α)첨가에 따라 결정되었다. 데이터는 삼중 항에서 수단±sem 으로 표시되었다., (F)hek293T 세포에서 24h 에 대해 TNF-α 로 처리 한 후 CRISPR/Cas9 로 CCL2 의 표적 삭제 효율. (G)거대 세포 바이러스(CMV),또는 SV40,또는 hek293T 세포에서 염기성 프로모터로부터의 루시퍼 라제 활성의 수준. 데이터는 삼중 항에서 수단±sem 으로 표시되었다. (H)CMV,또는 SV40,또는 hek293T 세포에서 염기성 프로모터에 의해 제어되는 루시퍼 라제 유전자의 표적 삭제 효율. (B–D,F,H)결실의 퍼센트는 접합부를 가로 질러 또는 결실 영역 내에서 프라이머를 사용하여 RT-PCR 에 의해 정량화되었다.,
의 식 CCL2 유전자에 의해 극적으로 유도 TNF-α(최대∼300-fold increase)in HEK293T cells(그림 3E)을 제공,좋은 모델을 조사하는지 여부를 CRISPR/Cas9 중재 대상으로 게놈 편집에 의해 영향을 받는 transcriptional activity. 관심의 효율성을 대상으로 삭제 CCL2 유전자 소재에 의해 영향을 받지 않으로 세포의 치료 TNF-α(그림 3 층),제안 녹음방송을 변경하지 않았 CRISPR/Cas9 중재 삭제합니다., 더 이 결과를 확인,우리 대상으로 외인성 기자의 유전자에 의해 구동되는 다양한 프로모터와 다른 강점,다른 transcriptional 활동이 될 수 있는 평가를 사용하여 루시페라아제 분석 결과(그림 3G). PCR 분석을 밝혔 유사한 효율적으로 삭제 HEK293T cells co-transfection 후 기자의 유전자를 함께 Cas9 및 gRNA 쌍이다(그림 3). 이 결과는 NHEJ 매개 수리가 다양한 정도의 전사 활성이 발생 함에도 불구하고 발생할 수 있음을 나타냅니다.,
DSBs 는 HDR 을 자극하여 손상된 부위를 동종 기증자로 매우 정밀하게 대체 할 수 있습니다. 표적화 된 게놈 DNA 치환을 얻기 위해,우리는 한 쌍의 gRNAs,Cas9 및 표적화 된 영역에 상 동성을 갖는 선형 공여체를 세포 내로 도입했다(그림 4A). 선형 공여체는 50bp 의 동종 서열을 갖는 프라이머로 PCR 증폭에 의해 얻어졌다. 이 동일한 기증자는 zfn 기반 HDR 복구 시스템(18)을 사용하여 성공적으로 삽입되었습니다., 시험의 타당성 CRISPR/Cas9 중재에 의하여 보충 HDR,우리는 우리를 대상으로 CCL2 소재로는 한 쌍의 gRNAs(#39#1854 년에 표시된 그림 3)과 기증자 베어링은 향상된 녹색형광단백질(EGFP)코딩 순서와 SV40poly(A)사이트(Figure4A;시퀀스와 위치에서 제시하는 보충 자료). 이 시스템을 사용하여 약 0.5%의 대상으로 세포 EGFP-긍정적인 반면,만 0.023%었 EGFP-에 긍정적인 조롱 transfection 세포(단 transfected 기증자)하 제어 세포(0.021%,없이 transfection)., 이어서 유동 세포 계측법에 의해 egfp 양성 세포를 분류 하였다. 사이트 별 통합은 동종 암과 대체 된 전체 영역 모두를 측면으로하는 두 쌍의 프라이머를 사용하여 PCR 에 의해 확인되었다. 그림에서와 같이 4B,우리가 관찰하고 예상한 대체 지역을 포함하는 전체 길이 EGFP 시퀀스와 동종 무기(생어 시퀀싱 결과는 다음과 같이 추가 소재). 내인성 야생형 대립 유전자도 검출되었으며(그림 4B),모든 대립 유전자가 표적화 된 것은 아니라는 것을 나타낸다., 또한,우리가 선택한 단일 클론에서 EGFP-긍정적인 세포 및 발견되는 모든 클론(6 6 조사)가 예상되는 통합(그림 4C)하지만,생 wild-type allele 도에서 발견 세 가지의 클론(그림 4C),제안하는 단 하나의 대립 된 대상에서 이러난다. 식 EGFP 에서 단백질을 대상으로 세포(EGFP-긍정적인 정 세포)었까지 규제에서 TNF-α 처리 계산에 의해 서쪽 오 점와 형광 활성화 셀 정렬(FACS)(그림 4,D and E)., 이러한 결과는 CRISPR/Cas9 시스템이 높은 효율과 정확도로 유전자/도메인 대체물을 만드는 데 사용될 수 있음을 입증했습니다.

(A)인간 세포에서 CRISPR/Cas9 를 사용하여 표적 유전자 치환 절차를 묘사하는 개략도., 의 효율성을 테스트하기 위해 타겟 유전자체,가이드 RNAs(gRNAs)하도록 설계되었는 삭제된 지역(에 의해 표시되는 사이트 1 과 2)의 CCL2 유전자와 대체하는 삭제된 지역으로 EGFP-polyA 카세트 기증자와 함께 팔로 짧은 지역의 homology. 제 1 부위 및 제 2 부위의 표적 부위는도 3 에 도시 된 CCL2 유전자 내에서#39 및#1854 이다. 동종 서열(50bp)은 삭제 사이트의 업스트림 및 다운 스트림에 불과합니다. 자세하게 위치 및 서열은 보충 자료에 제시되어있다., (B)crispr/Cas9 로의 표적 유전자 치환 효율은 HEK293T 세포에서 PCR 에 의해 결정되었다. CCL2 와 EGFP 사이의 접합부에 걸친 프라이머를 PCR 증폭에 사용 하였다. (C)단일 클론의 표적 유전자 치환에 대한 PCR 분석. (D,E)HEK293T 세포에서 TNF-α 첨가시 EGFP 단백질의 발현은(D)웨스턴 블롯 및(E)FACS 에 의해 결정되었다.
여기서 우리는 설명하는 간단하고 효율적인 방법에 대한 유전자 삭제를 사용하는 CRISPR/Cas9 시스템입니다., 우리는 증명하는 소개의 이 시스템으로 인 HEK293T cells,및 다른 사람의 유형 셀,유도 삭제 조각의 10kb 효율성과 함께 이르기까지 사이 11%68%에 따라,대상 시퀀스입니다. 게놈 세그먼트를 효율적이고 정확하게 삭제할 수있는 능력은 인간 세포에서 기능적 게놈 요소의 연구를 용이하게 할 것입니다. 이 접근법은 잠재적으로 모든 게놈 위치를 표적으로 삼는 데 사용될 수 있습니다.
CRISPR/Cas9 시스템의 특이성에 관한 우려가 있었다(19-21)., 규칙을 원치 않는 고기로 인해 대상에서 돌연변이,우리는 적어도 두 개의 서로 다른 쌍의 gRNAs 위해 사용될 각 대상 영역이다. 우리의 연구에서 여러 gRNA 쌍의 필요성은이 시스템의 단순성과 고효율을 감안할 때 큰 제한이 아니 었습니다. 동일한 지역을 대상으로 한 다른 쌍의 gRNAs 가 높은 효율로 작동했다는 것은 주목할 만하다(그림 2 와 4). 원치 않는 돌연변이를 피하기위한 또 다른 접근법은 이중 니카 제 방법을 사용하는 것입니다(22,23)., 우리는 또한 게놈 DNA 의 삭제를 생성하기 위해 이중 nickase 방법을 성공적으로 적용했지만 효율은 상당히 낮았다.
하는 것으로 알려져있다 복의 DNA DSBs 주로 중재에 의해 오류가 발생하기 쉬운 NHEJ,에서는 두 종료 처리 및 출혈이 함께 하는 방법에 자주 동행하여 뉴클레오티드 삽입과 삭제. 이러한 오류가 발생하기 쉬운 엔드 결합은 ZFNs 또는 TALENs 에 의해 생성 된 Dsb 의 수리에서 관찰되었습니다. 대조적으로,Cas9 와 2 개의 gRNAs 에 의해 생성 된 Dsb 의 수리는 매우 정확했다., 우리의 결과는 휴식이 끝 처리없이 직접 결찰되어 NHEJ 경로의 이전에 인정되지 않은 이점을 드러냄을 시사한다. 정확한 결찰을 초래하는 메커니즘은 여전히 결정되어야합니다. 하나의 가능성은 그 대상으로 삭제를 사용하여 Cas9 두 gRNAs 결과에 접속하는 인식하지 못하거나 원래의 gRNAs. 우리는 또한 분석 효율성에서 생성 indel 돌연변이를 위한 개별 gRNA 및 gRNA 쌍(#39#224gRNA 에서 도 3A)의 생어 시퀀싱 PCR amplicons 의(TA 복제)., 의 관심이,우리가 관찰의 효율성 indel 돌연변이를 위한 단일 gRNA 었고 매우 낮(9.5%,2 개의 21 클론을 대#39gRNA;5%,1 20 클론을 대#224gRNA). 그러나 gRNA 쌍은 indel 돌연변이(#39 및#224 에 대해 50%,20 클론 중 10 개)의 고효율을 생성했으며,이는 qPCR 을 사용한 분석 결과와 유사했다(52%,그림 3b). 우리는 단일 gRNA 가 종종 NHEJ 에 의해 정확하게 수리 될 분열 부위의 한 무딘 끝을 초래한다고 제안합니다. 따라서,돌연변이를 생성하는 효율은 한 쌍의 grna 보다 단일 gRNA 의 사용으로 훨씬 낮다.,
인정
이 작품은 국립 보건원(National Institute Of Health)의 보조금으로 지원되었습니다(No. DP1CA174421)및 C.-Z.C 에 W.M.Keck 재단,중국 국립 자연 과학 재단(No.81101481)및 상하이 의료 인재 양성 프로그램(No. XYQ2011048)To S.L.H. 이 논문은 NIH 공개 액세스 정책의 적용을받습니다.
경쟁 관심사
저자는 경쟁 관심사가 없다고 선언합니다.
보충 자료
이 논문에 수반되는 보충 자료를 보려면 저널 웹 사이트(www)를 방문하십시오.,future-science.com/doi/suppl/10.2144/000114196