Presis-genet sletting og erstatning ved å bruke CRISPR/Cas9 system i humane celler
Her viser vi at to guide RNAs kombinert med Cas9 effektivt generere DNA sletting av opp til 10 kb i humane celler i en prosess hvor reparasjon av slettingen er i stor grad oppnådd ved presis slutt å bli med. I tillegg gir vi data som viser at CRISPR/Cas9 system kan erstatte store genom fragmenter i nærvær av en lineær homologe reparasjon donor.,
bakteriell gruppert regelmessig interspaced kort palindromic gjentar/ CRISPR—forbundet (CRISPR/Cas) loci kode RNA-guidet immunforsvar som beskytter cellene mot invaderende virus og plasmider (1, 2). I Streptococcus pyogenes, type II CRISPR/Cas-systemer bruker en RNA-guidet endonuclease (RGEN), Cas9, for å katalysere site-specific spalting av mål-DNA-sekvenser., Målretting av Cas9 til bestemte genom nettsteder er mediert av en 20 nukleotid guide rekkefølge innen en tilknyttet CRISPR RNA (crRNA) og krever en trans-aktivere crRNA (tracrRNA) som rekrutter de crRNA i Cas9 komplekse (3). Anerkjennelse av spalting områder er bestemt av crRNA-DNA-base sammenkobling og en protospacer-tilstøtende motiv (PAM), en tre nukleotid-sekvensen (NGG) satt til komplementære DNA-regionen (4)., Det er bemerkelsesverdig at en enkelt guide RNA (gRNA) som etterligner tracrRNA-crRNA komplekset kan rekruttere Cas9 til målrettede genom nettsteder og generere dobbel-strandet pauser (DSBs) i DNA (5). Den CRISPR/Cas9 systemer har blitt tilpasset for site-specific genom redigering i ulike celletyper og organismer (6-12).
Genom redigering med CRISPR/Cas9 er igang med innføring av DSB på en målrettet genom locus ved hjelp av RNA-programmert RGEN. Dette er fulgt ved reparasjon av DSB gjennom enten homologi-anvist reparasjon (HDR) eller nonhomologous end-bli med (NHEJ)., I nærvær av et homologt reparasjon donor, den CRISPR/Cas9 systemet kan brukes til å generere presise og definert endringer og innsettinger på en målrettet locus gjennom HDR-prosessen. I fravær av et homologt reparasjon donor, enkelt DSBs generert av CRISPR/Cas9 er reparert gjennom utsatt for feil-NHEJ, noe som resulterer i innsetting eller sletting (indel) mutasjoner. Indel mutasjoner i koding exons kan innføre tidlig stopp codons eller frame-shift-mutasjoner, og dermed å deaktivere tilsvarende proteiner., Indel mutasjoner generert fra reparere en enkelt DSB kan ikke være nyttig i eksperimenter som tar sikte på å karakterisere den funksjonelle domener av protein-kodende gener eller for inaktivering av genomisk elementer som intergenic eller intronic regulatoriske sekvenser eller noncoding RNA gener. DNA-fragment slettinger i mål loci ville gi en avenue for å studere disse funksjonelle elementer. For dette formål, flere DSBs har blitt introdusert for å generere slettinger i Drosophila (12, 13), zebrafisk (14), og menneskelige celler (8), om enn med lav effektivitet., Målrettet genomisk DNA slettinger har også vært oppnådd ved hjelp av sink finger nuclease (ZFN) eller transkripsjon aktivator-som effector nuclease (TALEN) i humane celler (15-17). Imidlertid effektiviteten av disse metodene er generelt lave. I tillegg, ZFNs og TALENs være litt vanskelig og dyrt å utforme, utvikle og empirisk test i mobil sammenheng.
Her har vi undersøkt generasjon av fragment slettinger i humane celler catalyzed av CRISPR/Cas9 system. Vi viser at 2 gRNAs kombinert med Cas9 effektivt kan lage DNA sletting av opp til 10 kb., Av interesse, fant vi ut at reparasjon av dette sletting prosessen er i stor grad oppnådd ved presis slutt å bli med. Videre, målrettet sletting med CRISPR/Cas9 ser ut til å være uavhengig av transcriptional status på det aktuelle locus. Til slutt viser vi at CRISPR/ Cas9 systemet kan brukes til å erstatte store genom fragmenter i nærvær av en lineær homologe reparasjon donor.
Materiale og Metoder
Plasmider bygging
Den grunnleggende H1 arrangøren ble forsterket fra pLVTHM plasmider (Addgene, #12247, Cambridge, MA)., Oligonukleotider som inneholder endret H1 arrangøren og ryggraden i ønsket gRNA sekvenser med to BsaI steder ble syntetisert (PAN-Anlegget, Stanford University). Den resulterende full-lengde produkter ble forsterket ved PCR og klonet inn i pUC19 vektor. Ampicillin genet (amp) og H1 arrangøren i pUC19 vektor inneholder BsaI restriction enzym nettsteder, disse ble mutert (amp genet ble endret fra G1601C, som ikke endre aminosyresekvens; den H1 arrangøren ble endret fra GAGACC å GAGGACC) for å eliminere BsaI nettsteder., Protokoll for gRNA kloning er presentert i Supplerende Materiale. Alle målretting nettsteder sekvenser er presentert i Supplerende Tabell S1.
Celle kultur
HEK 293T, SK-Hep1, og HeLa-cellene ble dyrket i Dulbecco ‘s modified Eagle»s medium (DMEM) supplert med 10% føtalt bovint serum (FBS) (Hyclone, Logan, UT) og penicillin/streptomycin (penn/strep) (Invitrogen, Carlsbad, CA, usa). PC3 cellene ble dyrket i RPMI-1640 middels supplert med 10% FBS og penn/strep., For tumor nekrose faktor-α (TNF-α) stimulering, 293T celler ble behandlet med angitt konsentrasjoner av TNF-α(R&D Systemer, Minneapolis, MN). Cellene ble opprettholdt ved 37°C og 5% CO2 i en humidified inkubator.
Målrettet DNA-sletting
HEK 293T celler ble seedet i 12-bra plater på en tetthet på 100 000 celler per brønn. Etter 24 t, cellene ble kortvarig transfekte med 1 µg Cas9 plasmider (Addgene, #41815), 0.5 µg gRNA T1, og 0,5 µg gRNA T2 ved hjelp av plasmider Lipofectamine 2000 (Invitrogen) som per produsent»s protokoller., Genomisk DNA ble ekstrahert 48 t etter transfection ved hjelp av QuickExtract DNA-Ekstraksjon Løsning (Episenter Biotechnologies, Madison, WI). Vanlig PCR ble utført for å forsterke den målrettede regionen ved å bruke primere som flankerer målrettet regioner. Wild-type og avkortet genom fragmenter ble løst ved gel elektroforese. Real-time PCR (RT-PCR) ble utført for å kvantifisere prosent av slettingen ved å bruke primere over krysset eller innenfor sletting regionen. Den komparative Cq metoden ble brukt til å beregne uttrykket nivå i målområdet i forhold til en referanse regionen (ACTB locus)., Prosent av sletting i målområdet ble videre beregnet ved forholdet mellom målceller i forhold til kontroll celler. Alle primer sekvenser er oppført i Supplerende Tabell S2.
Målet sekvensering
Cellene ble høstet to dager etter transfection, og genomisk DNA ble ekstrahert ved hjelp av QuickExtract DNA-Ekstraksjon Løsning (Episenter Biotechnologies). PCR ble utført for å forsterke målretting regionen med genomisk DNA stammer fra celler, og amplikoner var dypt sekvensert ved MiSeq Personlige Sequencer (Illumina, San Diego, CA, usa).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Donor-sekvenser er presentert i Supplerende Materiale. De to 5′-de fleste sammenhengene er phosphorothioate (merket med en stjerne). Cellene i 6-bra plater ble kortvarig transfekte med 2.0 µg Cas9 plasmider, som er 0,8 µg gRNA T1 plasmider, som er 0,8 µg gRNA T2 plasmider, og 0,4 µg lineær donor ved hjelp av Lipofectamine 2000 (Invitrogen). På 48 t etter transfection, celler ble behandlet med 1 ng/mL TNF-α for 24 timer, og deretter GFP-positive celler ble sortert.
Luciferase assay
For luciferase assay, HEK 293T celler ble seedet i 96-brønns plater på en tetthet på 5000 celler per brønn., Etter 24 t, cellene ble kortvarig transfekte med 5 ng av pRL-TK Renilla luciferase reporter og 100 ng av luciferase reporter med cytomegalovirus (CMV), SV40 (Simian virus 40), eller basic arrangøren. Etter 48 t, luciferase aktivitet ble målt med dual-luciferase reporter analysen system (Promega).
Western blot
Proteiner ble separert ved sodium dodecyl sulfat—SIDE (SDS-PAGE) og overført til nitrocellulose membraner. Membraner ble blokkert med 5% nonfat melk og inkubert med GFP-antistoff (CST, #2555S, Danvers, MA)., Antigen-antistoff-komplekset ble oppdaget med forbedret chemiluminescence reagenser.
Resultater og diskusjon
Vi tilpasset den bakterielle type II CRISPR/ Cas9 system for å mutagenize genomisk DNA i humane celler. Den menneskelige codon-optimalisert versjon av S. pyogenes Cas9 protein lager en C-terminus SV40 kjernefysiske lokalisering signalet ble uttrykt ved hjelp av et tidligere beskrevet system (6). Å direkte Cas9 spalting til ønsket sekvens, vi uttrykte crRNA-tracrRNA fusion transkripsjoner, heretter referert til som guide RNAs (gRNAs), fra en modifisert menneskelige H1-polymerase III arrangøren., 3′ enden av H1 arrangøren ble endret for å tillate transkripsjon av gRNAs som begynner med noen nukleotid. Begrenset bare av kravet om at 20 bp crRNA mål følges av PAM sekvens, NGG (hvor N er et nukleotid) denne metoden kan i prinsippet brukes til å målrette eventuelle genom sted som har form N20NGG. For å forenkle kloning av gRNA uttrykk vektor, vi brukte en Type IIs restriction enzym, BsaI. Dette nødvendig for syntese av en 24 bp oligonukleotid inneholder en region av komplementær til mål område på DNA., Enkel og effektiv protokoll for kloning av gRNA uttrykk vektor (Figur 1A) er beskrevet i detalj i den Supplerende Materiale.
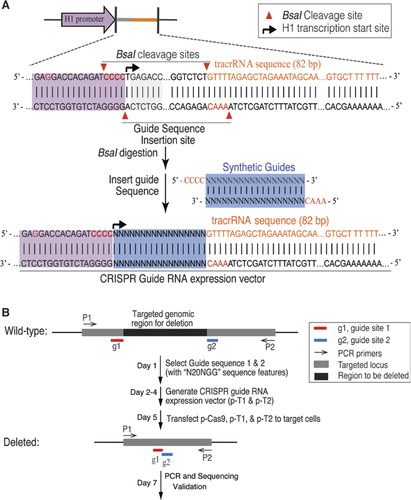
(A) Utforming av veiledningen RNA (gRNA) uttrykk vektor. Vector var designet for å produsere gRNA transkripsjoner med en syntetisk gRNA smeltet til trans-aktivering av RNA/CRISPR RNA (tracrRNA)., Den H1 arrangøren ble endret for å eliminere intern type IIS restriction enzym BsaI området ved å endre GAGACC i H1 arrangøren å GAGGACC. En BsaI nettstedet ble introdusert for å skape kloning nettsteder for gRNA og tracrRNA fusions ved innsetting av syntetiske oligonukleotid tomannsboliger kompatibel med overheng. 3′ enden av H1 arrangøren ble endret til å være CCACAGATCCCC til rette for transkripsjon av gRNAs med noen nukleotid i 5′ – enden. (B) trinn av målrettede genet sletting med CRISPR/Cas9.,
For sletting av et stort segment av genomisk DNA, har vi brukt et par gRNAs mot målrettede locus (Figur 1B). To mål områder med mønster N20NGG ble valgt på grensen til målområdet. Effektiviteten av målrettede sletting veiledet av ulike kombinasjoner av gRNA parene var bestemt av PCR-analyser ved bruk av primere som flankerer målrettet regioner. Wild-type og avkortet genom fragmenter ble løst ved gel elektroforese. For å unngå PCR-amplifikasjon bias, prosent sletting ble kvantifisert ved RT-PCR bruke en primer par., Primerne ble utformet over sletting junction (en primer utenfor sletting regionen, andre primer innenfor sletting region) eller innenfor sletting regioner (både primere finne innenfor sletting region). Dermed ble det bare en eneste bandet som er amplifisert med primeren par for både målrettet celler og kontroll celler. Vi beregnet prosent sletting ved å sammenligne den relative mengden av PCR-produkter (målceller versus kontroll-celler) forsterket av den samme primerpar. Målrettet slettinger ble ytterligere bekreftet ved sekvensering.,
for Å vurdere hvordan gRNA par kan påvirke etterfølgende reparasjon og generering av slettinger, må vi først utformet sett av gRNAs rettet mot den menneskelige CDC42 genom locus og atskilt av avstander fra ca 200 til 10 000 bp (Figur 2A og Supplerende Tabell S1). Vi har da vurdert muligheten av hver gRNA par for å generere slettinger i menneskelig HEK 293T celler i nærvær av Cas9. Robust effektiviteten av NHEJ-basert slettinger (opptil 68%) ble bekreftet av qPCR (Figur 2B–2D)., Selv for sletting av en 10 kb genom regionen, fikk vi målretting priser på 16% til 28%, avhengig av gRNA par. Dette RNA-mediert prosessen var rask, med den første påviselige sletting vises ca 12 h post-transfection (Supplerende Figur S1). Systemet var effektivt i en rekke celletyper, inkludert: PC3, SK-Hep1, og HeLa-celler (Supplerende Figur S2).

(A) Skjematisk diagram som viser plasseringen av guide RNAs (gRNAs) rettet mot CDC42 locus. (B–D) effektiviteten av målrettede sletting med CRISPR/Cas9 ble bestemt ved PCR i HEK 293T celler. Primere utenfor forventet sletting regioner ble brukt. Prosent av slettingen ble kvantifisert ved RT-PCR med primere over krysset eller innenfor sletting regionen. (E–F) effektivitet og presisjon målrettet sletting i CDC42 ble bekreftet av (E) Sanger-sekvensering og (F) høy gjennomstrømning sekvensering analyser. PCR-produktet inneholder bare sletting amplikon ble beriket for sekvensering.,
Slettinger ble ytterligere bekreftet ved sekvensering av PCR-produkter som spenner over forventet spalting nettsteder. Sanger-sekvensering viste sletting veikryss resulterte fra den nøyaktige ligation av den butte-endte DSBs opprettet av Cas9; hver DSB skjedde nøyaktig 3 bp oppstrøms av PAM sekvens (Figur 2E og Supplerende Figur S3). Vi har også brukt dyp sekvensering av sletting amplikoner for å vurdere nøyaktigheten av sletting effektivitet; i om lag 80% av lyder, målrettet DSBs var perfekt reparert (Figur 2F).,
Vi en oppsummering av disse funnene av testing gRNA par utformet for å slette fragmenter fra et genom locus inneholder microRNA miR-21 gen i HEK 293T celler. To gRNAs var utformet for å målrette mot grensene av miR-21 hårnål (Supplerende Figur S4). Sletting effektivitet var 38% følgende transfection med to gRNAs og Cas9, som måles ved hjelp av en PCR-analyse (Supplerende Figur S4B). Dyp sekvensering bekreftet sletting skjedde nøyaktig som forventet (Supplerende Figur S4C).,
for Å undersøke om de CRISPR/ Cas9-mediert genet sletting system er påvirket av transcriptional staten målrettet gener, genkode den chemokine (C-C-motif) ligand 2 (CCL2) var målrettet. CCL2 er en liten cytokin som hører til CC chemokine familie; CCL2 genet er et mål av NF-kB-signalering. Vi tilfeldig valgt åtte mål steder i 5′ – og 3′ endene av CCL2 gen-loci (Figur 3A). Vi oppnådd robust og effektiv sletting av ulike regioner av genet ved hjelp av Cas9 og ulike par av gRNAs i HEK 293T celler (Figur 3B–3D).,

(A) Skjematisk diagram som viser plasseringen av guide RNAs (gRNAs) rettet mot CCL2 locus. (B–D) effektiviteten av målrettede sletting av CCL2 med CRISPR/Cas9 i HEK 293T celler. (E) CCL2 mRNA-nivå ble bestemt ved tumor nekrose faktor-α (TNF-α) tillegg ved kvantitativ RT-PCR analyser i HEK 293T celler. Data ble vist med midler ± sem i triplikat., (F) effektiviteten av målrettede sletting av CCL2 med CRISPR/Cas9 etter behandling med TNF-α for 24 t i HEK 293T celler. (G) nivå av luciferase aktivitet fra cytomegalovirus (CMV), eller SV40, eller en grunnleggende arrangøren i HEK 293T celler. Data ble vist med midler ± sem i triplikat. (H) Effektivitet av målrettede sletting av luciferase genet kontrolleres av CMV, eller SV40, eller en grunnleggende arrangøren i HEK 293T celler. (B–D,F,H) prosent av slettingen ble kvantifisert ved RT-PCR med primere over krysset eller innenfor sletting regionen.,
Uttrykk for CCL2 genet var dramatisk indusert av TNF-α (opp til ∼300-fold økning) i HEK 293T celler (Figur 3E), som gir en god modell for å undersøke om de CRISPR/ Cas9-mediert målrettet genom redigering er påvirket av transcriptional aktivitet. Av interesse er effektiviteten av målrettede sletting av CCL2 gen-loci ble ikke påvirket av behandling av celler med TNF-α(Figur 3F), noe som tyder på at transkripsjon ikke endre CRISPR/Cas9-mediert sletting., For å ytterligere bekrefte dette resultatet, vi rettet en eksogen reporter gen drevet av ulike arrangører med forskjellige styrker, hvor de forskjellige transcriptional aktiviteter kan bli vurdert ved hjelp av en luciferase assay (Figur 3G). PCR-analyser avslørte lignende effektivitet i målrettet slettinger i HEK 293T celler etter co-transfection av reporter gen sammen med Cas9 og gRNA par (Figur 3H). Dette resultatet indikerer NHEJ-mediert reparasjon kan skje til tross for at forekomsten av varierende grader av transcriptional aktivitet.,
DSBs kan stimulere HDR for å aktivere nøyaktig utskifting av skadede området med et homologt donor. For å oppnå målrettet genomisk DNA erstatning, har vi introdusert et par av gRNAs, Cas9, og en lineær donor med homologi til den målrettede regionen inn i cellene (Figur 4A). Den lineære donor ble innhentet av PCR-amplifikasjon med primere med 50 bp av homologe sekvenser. Denne samme donor er riktig satt inn ved hjelp av en ZFN-baserte HDR-repair system (18)., For å teste muligheten av CRISPR/ Cas9-mediert utskifting av HDR, vi målrettet CCL2 locus med et par av gRNAs (#39 #1854 vist i Figur 3) og en donor med forbedret grønt fluorescerende protein (EGFP) kodende sekvens og SV40 poly(A) området (Figur 4A; sekvenser og posisjoner er presentert i Supplerende Materiale). Ved hjelp av dette systemet, ca 0,5% av målrettede celler ble EGFP-positive, mens bare 0.023% var EGFP-positive i mock transfection celler (bare transfekte med donor), som var lik for å kontrollere celler (0.021%, uten transfection)., Den EGFP-positive cellene ble deretter sortert ved flowcytometri. Site-specific integrering ble bekreftet av PCR ved hjelp av to par av primere som flankerer både homologe armer og hele erstattet regionen. Som vist i Figur 4B, observerte vi forventet erstattet området som inneholder full lengde EGFP rekkefølge og homologe armer (Sanger-sekvensering resultatet vises i den Supplerende Materiale). Den endogene wild-type allelet ble også påvist (Figur 4B), noe som indikerer at ikke alle alleler er målrettet., Videre har vi valgt ett kloner fra EGFP-positive celler, og fant at alle klonene (6 av 6 undersøkt) hadde forventet integrasjon (Figur 4C), men den endogene wild-type allelet ble også påvist i tre av kloner (Figur 4C), noe som tyder på at bare ett allel var målrettet i de kloner. Uttrykk for EGFP protein i målrettet celler (EGFP-positive sortert celler) ble opp-regulert på TNF-α-behandling som vurderes av Western blot og fluorescens aktivert celle-sortering (FACS) (Figur 4, D og E)., Disse resultatene viste at CRISPR/Cas9 systemet kan brukes til å lage genet/domene erstatninger med høy effektivitet og nøyaktighet.

(A) Skjematisk diagram som viser prosedyren for målrettet genet erstatning ved hjelp av CRISPR/Cas9 i humane celler., For å teste effekten av målrettede genet erstatning, guide RNAs (gRNAs) ble utviklet for å slette den angitte regionen (merket med nettsteder 1 og 2) av CCL2 gen og å erstatte slettet regionen med EGFP-polyA kassett donor med armer med kort regioner av homologi. De målrettede stedene i område 1 og område 2 er #39 #1854 i CCL2 genet vist i Figur 3. De homologe sekvenser (50 bp) er like oppstrøms og nedstrøms av sletting nettsteder. Posisjoner og sekvenser i detalj er presentert i Supplerende Materiale., (B) effektiviteten av målrettede genet erstatning med CRISPR/Cas9 ble bestemt ved PCR i HEK 293T celler. Primere som spenner over veikryss mellom CCL2 og EGFP ble brukt for PCR-amplifikasjon. (C) PCR-analyse for målrettet genet erstatning for enkelt-kloner. (D,E) Uttrykk for EGFP protein på TNF-α tillegg i HEK 293T celler ble bestemt ved (D) Western blot og (E) FACS.
Her beskriver vi en enkel og effektiv tilnærming for gene sletting ved hjelp av CRISPR/Cas9 system., Vi har vist at innføringen av dette systemet i menneskelig HEK 293T cellene, og andre menneskelige celletyper, indusert sletting av fragmenter opp til 10 kb med effektiviteten varierer mellom 11% og 68%, avhengig av målrettede rekkefølge. Muligheten til å effektivt og presist slett genom segmenter vil legge til rette for studier av funksjonell genomisk elementer i humane celler. Denne tilnærmingen kan være potensielt brukes til å målrette eventuelle genom loci.
Det har vært bekymring om spesifisitet av CRISPR/Cas9 system (19-21)., For å utelukke uønskede phenotypes på grunn av off-target mutasjoner, foreslår vi at minst to forskjellige par av gRNAs brukes for hvert mål regionen. I vår studie, er det behov for flere gRNA par var ikke en stor begrensning, gitt enkelhet og svært effektiviteten av dette systemet. Det er bemerkelsesverdig at ulike par av gRNAs målrettet i samme region jobbet med høy effektivitet (Figur 2 og 4). En annen tilnærming for å unngå uønskede mutasjoner er bruk av doble nickase metode (22, 23)., Vi har også med hell brukes dobbel nickase metode for å generere sletting av genomisk DNA, men effektiviteten var betydelig lavere.
Det er kjent at reparasjon av DNA DSBs er i stor grad påvirket av utsatt for feil-NHEJ, der de to endene er behandlet og ligated sammen på en måte som er ofte ledsaget av nukleotid innsettinger og slettinger. Slike utsatt for feil ende å bli observert i reparasjon av DSBs opprettet av ZFNs eller TALENs. I kontrast, reparasjon av DSBs generert av Cas9 og to gRNAs var svært nøyaktig., Våre resultater tyder på at pausene er direkte ligated uten ende behandling, avslører et tidligere verdsatt nytte av NHEJ veien. Mekanismen som resulterer i nøyaktig ligations er fortsatt ikke bestemt. En mulighet er at målrettede sletting ved hjelp av Cas9 og to gRNAs resultater i et knutepunkt som ikke er anerkjent av noen av de opprinnelige gRNAs. Vi har også analysert effektivitet i å generere indel mutasjoner for den enkelte gRNA og gRNA par (#39 #224 gRNA i Figur 3A) med Sanger-sekvensering av PCR-amplikoner (TA-kloning)., Av interesse, observerte vi at effektiviteten av å generere indel mutasjoner for enkelt gRNA var ganske lav (9.5%, 2 av 21 kloner for #39 gRNA; 5%, 1 av 20 kloner for #224 gRNA). Men gRNA par generert høy effektivitet av indel mutasjoner (50%, 10 av 20 kloner for #39 #224), som var lik den analysen ved bruk av qPCR (52%, Figur 3B). Vi foreslår at en enkelt gRNA resulterer ofte i en butt ende av spalting nettstedet vil være nettopp reparert av NHEJ. Dermed effektiviteten av å generere mutasjon er mye lavere med bruk av et enkelt gRNA enn et par gRNAs.,
Erkjennelsene
Dette arbeidet ble støttet av midler fra National Institute of Health (Nr. DP1CA174421) og W. M. Keck foundation til C. Z. C, og National Natural Science Foundation of China (Nr 81101481) og Shanghai Medisinsk Talent Training Program (Nr. XYQ2011048) til S. L. H. Dette papiret er underlagt NIH Public Access-Policy.
Konkurrerende interesser
forfatterne erklærer ingen konkurrerende interesser.
Supplerende data
for Å vise den supplerende data som følge av dette papiret vennligst besøk tidsskriftet hjemmeside: www.,future-science.com/doi/suppl/10.2144/000114196