Precyzyjna delecja genu i wymiana za pomocą systemu CRISPR / Cas9 w komórkach ludzkich
tutaj pokazujemy, że dwa przewodniki RNA w połączeniu z Cas9 skutecznie generują delecje DNA do 10 kb w komórkach ludzkich w procesie, w którym naprawa delecji jest w dużej mierze dokonywana przez precyzyjne łączenie końcowe. Ponadto dostarczamy dane pokazujące, że system CRISPR / Cas9 może zastąpić duże fragmenty genomu w obecności liniowego homologicznego dawcy naprawczego.,
bakteryjne klastry regularnie przeplatane krótkie powtórzenia palindromiczne / CRISPR-associated (CRISPR / Cas) loci kodują systemy odpornościowe kierowane RNA, które chronią komórki przed inwazyjnymi wirusami i plazmidami (1, 2). W Streptococcus pyogenes, typ II CRISPR / Cas systemy używają RNA-guided endonuclease (rgen), Cas9, katalizować site-specific rozszczepienie docelowych sekwencji DNA., Kierowanie Cas9 do specyficznych miejsc genomicznych jest pośredniczone przez 20 nukleotydową sekwencję przewodnika w powiązanym CRISPR RNA (crRNA) i wymaga trans-aktywującego crRNA (tracrRNA), który rekrutuje crRNA do kompleksu Cas9 (3). Rozpoznawanie miejsc rozszczepienia jest determinowane przez parowanie zasad crRNA-DNA i motyw przylegający do protospacera( PAM), sekwencję trzech nukleotydów (NGG) zestawioną z regionem komplementarnym DNA (4)., Należy zauważyć, że pojedynczy przewodnik RNA (gRNA) naśladujący kompleks tracrRNA-crRNA może rekrutować Cas9 do docelowych miejsc genomicznych i generować dwuniciowe przerwy (DSBs) w DNA (5). Systemy CRISPR / Cas9 zostały przystosowane do edycji genomu site-specific w różnych typach komórek i organizmach (6-12).
edycja genomu z CRISPR / Cas9 jest inicjowana z wprowadzeniem DSB w docelowym genomowym locus przy użyciu RNA-programmed rgen. Następnie następuje naprawa DSB poprzez naprawę ukierunkowaną homologicznie (HDR) lub niehomologiczne łączenie końcowe (NHEJ)., W obecności homologicznego dawcy napraw, system CRISPR/Cas9 może być używany do generowania precyzyjnych i zdefiniowanych modyfikacji i wstawek w docelowym miejscu w procesie HDR. W przypadku braku homologicznego dawcy napraw, pojedyncze Dsb generowane przez CRISPR / Cas9 są naprawiane przez podatny na błędy NHEJ, co powoduje wstawianie lub delecję (indel) mutacje. Mutacje Indel w kodujących egzonach mogą wprowadzać przedwczesne kodony stop lub mutacje frame-shift, inaktywując w ten sposób odpowiednie białka., Mutacje Indel generowane przez naprawę pojedynczego DSB mogą nie być użyteczne w eksperymentach mających na celu scharakteryzowanie funkcjonalnych domen genów kodujących białka lub inaktywację elementów genomowych, takich jak intergeniczne lub introniczne sekwencje regulacyjne lub niekodujące geny RNA. Delecje fragmentów DNA w docelowych loci dałyby możliwość zbadania tych funkcjonalnych elementów. W tym celu wprowadzono wiele DSB do generowania delecji w Drosophila (12, 13), danio pręgowanym (14) i komórkach ludzkich (8), aczkolwiek z niską skutecznością., Celowane delecje genomowe DNA zostały również osiągnięte przy użyciu nukleazy palca cynkowego (ZFN) lub nukleazy efektorowej aktywatora transkrypcyjnego (TALEN) w komórkach ludzkich (15-17). Jednak skuteczność tych podejść jest na ogół niska. Ponadto Zfns i TALENs pozostają nieco trudne i kosztowne w projektowaniu, rozwijaniu i empirycznie testowaniu w kontekście komórkowym.
tutaj zbadaliśmy generowanie delecji fragmentów w komórkach ludzkich katalizowanych przez system CRISPR/Cas9. Pokazujemy, że 2 grna w połączeniu z Cas9 mogą skutecznie tworzyć delecje DNA o wielkości do 10 kb., Co ciekawe, odkryliśmy, że naprawa tego procesu usuwania jest w dużej mierze dokonywana przez precyzyjne łączenie końcowe. Co więcej, celowe usuwanie za pomocą CRISPR / Cas9 wydaje się być niezależne od statusu transkrypcyjnego docelowego locus. Wreszcie, pokazujemy, że system CRISPR/ Cas9 może być używany do zastąpienia dużych fragmentów genomowych w obecności liniowego homologicznego dawcy naprawy.
materiał i metody
Budowa plazmidu
podstawowy promotor H1 został wzmocniony z plazmidu pLVTHM (Addgene, #12247, Cambridge, MA)., Zsyntetyzowano oligonukleotydy zawierające zmodyfikowany promotor H1 i szkielet pożądanych sekwencji gRNA z dwoma miejscami BsaI (Ośrodek PAN, Stanford University). Uzyskane produkty pełnej długości zostały wzmocnione przez PCR i sklonowane do wektora pUC19. Gen ampicyliny (amp) i promotor H1 w wektorze pUC19 zawierają miejsca enzymu restrykcyjnego BsaI; zostały one zmutowane (Gen amp zmieniono z G1601C, co nie zmienia sekwencji aminokwasów; promotor H1 zmieniono z Gagacc na GAGGACC) w celu wyeliminowania miejsc BsaI., Protokół klonowania gRNA przedstawiono w materiale uzupełniającym. Wszystkie sekwencje stron docelowych przedstawiono w dodatkowej tabeli S1.
hodowla komórkowa
komórki HEK 293t, SK-Hep1 i hela hodowano w zmodyfikowanej pożywce Dulbecco”s modified Eagle”(Dmem) uzupełnionej 10% surowicą bydlęcą płodu (FBS) (Hyclone, Logan, UT) i penicyliną/streptomycyną (Pen / strep) (Invitrogen, Carlsbad, CA). Komórki PC3 hodowano w pożywce RPMI-1640 uzupełnionej 10% FBS i Pen/strep., W celu stymulacji czynnika martwicy nowotworu α (TNF-α), komórki 293t były leczone wskazanymi stężeniami TNF-α (R&D, Minneapolis, MN). Komórki utrzymywano w temperaturze 37°C i 5% CO2 w nawilżonym inkubatorze.
celowana delecja DNA
komórki HEK 293T zostały zaszczepione w płytkach 12-studziennych o gęstości 100 000 komórek na studzienkę. Po 24 godzinach komórki zostały przejściowo przetoczone 1 µg plazmidu Cas9 (Addgene, #41815), 0,5 µg gRNA T1 i 0,5 µg gRNA T2 przy użyciu Lipofektaminy 2000 (Invitrogen) zgodnie z protokołami producenta., Genomowe DNA ekstrahowano 48 godzin po transfekcji za pomocą roztworu do ekstrakcji QUICKEXTRACT DNA (Epicentre Biotechnologies, Madison, WI). Common PCR przeprowadzono w celu wzmocnienia docelowego regionu za pomocą podkładów flankujących docelowe regiony. Dzikie i okrojone fragmenty genomowe zostały rozwiązane za pomocą elektroforezy żelowej. PCR w czasie rzeczywistym (RT-PCR) wykonano w celu określenia procentowego udziału delecji przy użyciu podkładów w poprzek węzła lub w obrębie regionu delecji. Porównawcza metoda Cq została użyta do obliczenia poziomu ekspresji regionu docelowego w stosunku do regionu odniesienia (actb locus)., Procent delecji w regionie docelowym był dalej obliczany przez stosunek komórek docelowych w stosunku do komórek kontrolnych. Wszystkie sekwencje podkładu są wymienione w dodatkowej tabeli S2.
sekwencjonowanie docelowe
komórki pobrano dwa dni po transfekcji, a genomowe DNA ekstrahowano za pomocą roztworu do ekstrakcji DNA QuickExtract (epicentrum Biotechnologie). PCR przeprowadzono w celu amplifikacji regionu docelowego z genomicznym DNA pochodzącym z komórek, a amplikony zostały głęboko zsekwencjonowane przez osobisty sekwencer MiSeq (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Sekwencje dawców przedstawiono w materiale uzupełniającym. Dwa wiązania o długości 5’to fosforotionian (oznaczany gwiazdkami). Komórki w płytkach 6-studziennych transfekowano przejściowo 2,0 µg plazmidu Cas9, 0,8 µg plazmidu gRNA T1, 0,8 µg plazmidu gRNA T2 i 0,4 µg dawcy liniowego z zastosowaniem Lipofektaminy 2000 (Invitrogen). Po 48 h po transfekcji komórki były leczone 1 ng / mL TNF-α przez 24 h, a następnie sortowano komórki GFP-dodatnie.
test Lucyferazy
w przypadku testu lucyferazy komórki HEK 293T zostały zaszczepione w płytkach 96-studziennych o gęstości 5000 komórek na studzienkę., Po 24 godzinach komórki zostały przejściowo przetoczone 5 ng pRL-TK renilla luciferase reporter i 100 ng luciferase reporter z cytomegalowirusem (CMV), SV40 (Simian virus 40) lub promotorem podstawowym. Po 48 godzinach aktywność lucyferazy oznaczano za pomocą systemu dual luciferase reporter assay system (Promega).
Western blot
białka zostały oddzielone przez siarczan dodecylu sodu—PAGE (SDS-PAGE) i przeniesione do błon nitrocelulozowych. Membrany zostały zablokowane 5% mlekiem beztłuszczowym i inkubowane przeciwciałem GFP (CST, #2555S, Danvers, MA)., Kompleks antygen-przeciwciało wykryto za pomocą wzmocnionych odczynników chemiluminescencyjnych.
wyniki i dyskusja
zaadaptowaliśmy bakteryjny system CRISPR / Cas9 typu II do mutagenizacji genomowego DNA w komórkach ludzkich. Zmodyfikowana przez kodon ludzki wersja białka S. pyogenes Cas9 z sygnałem lokalizacji jądrowej C-terminus SV40 została wyrażona przy użyciu wcześniej opisanego układu (6). Aby skierować rozszczepienie Cas9 do pożądanej sekwencji, wyraziliśmy transkrypty fuzji crRNA-tracrRNA, zwane dalej przewodnikiem RNAs (gRNAs), ze zmodyfikowanego promotora ludzkiej polimerazy H1 III., Koniec 3 ' promotora H1 został zmodyfikowany, aby umożliwić transkrypcję grna zaczynającą się od dowolnego nukleotydu. Ograniczone jedynie wymogiem, że 20 bp crrna docelowego być po sekwencji PAM, NGG (gdzie N jest dowolny nukleotyd), podejście to może w zasadzie być używany do kierowania dowolnej lokalizacji genomowej, która ma postać N20NGG. Aby ułatwić klonowanie wektora ekspresji gRNA, użyliśmy enzymu restrykcyjnego typu IIS, BsaI. Wymagało to syntezy oligonukleotydu 24 bp zawierającego region komplementarności do miejsca docelowego w DNA., Prosty i efektywny protokół klonowania wektora ekspresji gRNA (rysunek 1A) jest szczegółowo opisany w materiale uzupełniającym.
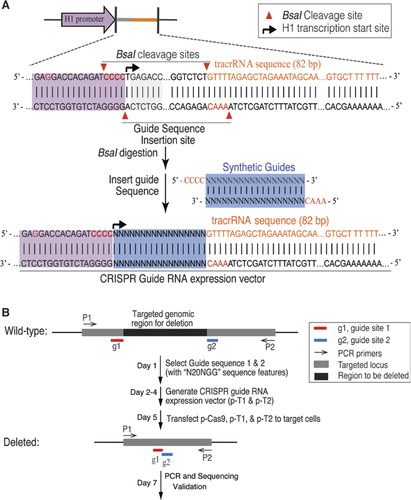
(A) projektowanie wektora ekspresji RNA (gRNA). Wektor został zaprojektowany do wytwarzania transkryptów gRNA z syntetycznym gRNA połączonym z trans-aktywującym RNA / CRISPR RNA (tracrRNA)., Promotor H1 został zmodyfikowany w celu wyeliminowania wewnętrznego enzymu restrykcyjnego BsaI typu IIS poprzez zmianę gagacc w promotorze H1 NA GAGGACC. Miejsce BsaI zostało wprowadzone w celu stworzenia miejsc klonowania fuzji gRNA i tracrRNA poprzez wstawienie syntetycznych dupleksów oligonukleotydów z Kompatybilnymi nawisami. Koniec 3′ promotora H1 został zmodyfikowany na CCACAGATCCC, aby ułatwić transkrypcję grna z dowolnym nukleotydem na końcu 5′. B) etapy ukierunkowanej delecji genów za pomocą CRISPR / Cas9.,
do usunięcia dużego segmentu genomowego DNA użyliśmy pary grna przeciwko docelowemu locus (rysunek 1B). Dwa obiekty docelowe o wzorze N20NGG zostały wybrane na granicy regionu docelowego. Efektywność celowanej delecji, opartej na różnych kombinacjach par gRNA, została określona za pomocą analiz PCR z użyciem podkładów flankujących docelowe regiony. Dzikie i okrojone fragmenty genomowe zostały rozwiązane za pomocą elektroforezy żelowej. Aby uniknąć błędu amplifikacji PCR, procentową delecję oznaczono metodą RT-PCR przy użyciu jednej pary podkładów., Podkłady zostały zaprojektowane w poprzek skrzyżowania delecji (jeden podkład poza regionem delecji, drugi podkład w regionie delecji) lub w regionach delecji (oba podkłady znajdują się w regionie delecji). W ten sposób tylko pojedyncze pasmo jest wzmacniane parą podkładu zarówno dla komórek docelowych, jak i komórek kontrolnych. Obliczyliśmy procentową delecję, porównując względną ilość produktów PCR (komórki docelowe w porównaniu z komórkami kontrolnymi) wzmocnionych przez tę samą parę podkładów. Celowe delecje były dodatkowo weryfikowane przez sekwencjonowanie.,
aby ocenić, w jaki sposób pary gRNA mogą wpływać na późniejszą naprawę i generowanie delecji, najpierw zaprojektowaliśmy zestawy grna skierowane przeciwko ludzkiemu genomowemu locus CDC42 i oddzielone odległościami od około 200 do 10 000 bp (rysunek 2A i dodatkowa tabela S1). Następnie oceniliśmy zdolność każdej pary gRNA do generowania delecji w ludzkich komórkach HEK 293T w obecności Cas9. Wysoka skuteczność delecji opartych na NHEJ (do 68%) została potwierdzona przez qPCR (rysunek 2B–2D)., Nawet dla delecji 10 kb genomic region, otrzymaliśmy targetowanie stawki od 16% do 28%, w zależności od gRNA para. Ten proces edycji za pośrednictwem RNA był szybki, a pierwsza wykrywalna delecja pojawiła się około 12 godzin po transfekcji (dodatkowa rycina S1). System był skuteczny w różnych typach komórek, w tym: PC3, SK-Hep1 i komórki HeLa (rysunek dodatkowy S2).

(A) Schemat przedstawiający lokalizacje guide RNAs (gRNAs) kierujące locus CDC42. (B–D) skuteczność ukierunkowanej delecji z CRISPR/Cas9 została określona przez PCR w komórkach HEK 293T. Zastosowano podkłady poza przewidywanymi regionami delecji. Procent delecji określono ilościowo za pomocą RT-PCR przy użyciu podkładów w poprzek połączenia lub w obrębie regionu delecji. (E–F) skuteczność i precyzja ukierunkowanej delecji w CDC42 została potwierdzona przez (E) sekwencjonowanie Sangera i (F) analizy sekwencjonowania o wysokiej przepustowości. Produkt PCR zawierający tylko amplikon delecji został wzbogacony do sekwencjonowania.,
delecje zostały dodatkowo potwierdzone przez sekwencjonowanie produktów PCR obejmujących spodziewane miejsca rozszczepienia. Sekwencjonowanie Sangera wykazało delecje wynikające z precyzyjnego ligowania tępo zakończonych DSBs utworzonych przez Cas9; każdy DSB wystąpił dokładnie 3 bp przed sekwencją PAM(rysunek 2e i rysunek S3). Wykorzystaliśmy również głębokie sekwencjonowanie amplikonów delecji do oceny dokładności skuteczności delecji; w około 80% odczytów docelowe Dsb zostały doskonale naprawione (rysunek 2F).,
podsumowaliśmy te odkrycia, testując pary gRNA mające na celu usunięcie fragmentów z genomowego locus zawierającego Gen microRNA miR-21 w komórkach HEK 293T. Dwa grna zostały zaprojektowane do celowania w granice mir-21 hairpin (rysunek dodatkowy S4). Skuteczność delecji wynosiła 38% po przetoczeniu z użyciem dwóch grna i Cas9, mierzonych metodą PCR (dodatkowa rycina S4B). Głębokie sekwencjonowanie potwierdziło, że delecja nastąpiła dokładnie zgodnie z oczekiwaniami (rysunek dodatkowy S4C).,
aby zbadać, czy system delecji genów za pośrednictwem CRISPR / Cas9 ma wpływ na stan transkrypcyjny docelowych genów, gen kodujący chemokinę (Motyw C-C) ligand 2 (CCL2) został ukierunkowany. CCL2 jest małą cytokiną należącą do rodziny chemokin CC; Gen CCL2 jest celem sygnalizacji NF-kB. Losowo wybraliśmy osiem miejsc docelowych zlokalizowanych na końcach 5 'I 3′ locus genu CCL2 (rysunek 3A). Osiągnęliśmy solidną i skuteczną delecję różnych regionów genu za pomocą Cas9 i różnych par grna w komórkach HEK 293T (rysunek 3B–3D).,

(A) Schemat przedstawiający lokalizacje guide RNAs (gRNAs) celujących w locus CCL2. (B–D) skuteczność ukierunkowanej delecji CCL2 z CRISPR / Cas9 w komórkach HEK 293T. (E) poziomy mRNA CCL2 oznaczano po dodaniu czynnika martwicy nowotworu α (TNF-α) za pomocą ilościowych analiz RT-PCR w komórkach HEK 293T. Dane pokazano za pomocą średniej ± sem w trzech egzemplarzach., F) skuteczność ukierunkowanej delecji CCL2 z CRISPR / Cas9 po leczeniu TNF-α przez 24 h w komórkach HEK 293T. G) poziom aktywności lucyferazy wywołanej wirusem cytomegalii (CMV) lub SV40 lub promotorem podstawowym w komórkach HEK 293T. Dane pokazano za pomocą średniej ± sem w trzech egzemplarzach. H) skuteczność ukierunkowanej delecji genu lucyferazy kontrolowanego przez CMV lub SV40 lub promotora podstawowego w komórkach HEK 293T. (B–D,F,H) procent delecji określono ilościowo za pomocą RT-PCR przy użyciu podkładów w poprzek połączenia lub w obrębie regionu delecji.,
ekspresja genu CCL2 została dramatycznie indukowana przez TNF-α (do ∼300-krotnego wzrostu) w komórkach HEK 293t (rysunek 3E), zapewniając dobry model do zbadania, czy edycja genomu ukierunkowanego za pośrednictwem CRISPR / Cas9 ma wpływ na aktywność transkrypcyjną. Co ciekawe, skuteczność ukierunkowanej delecji locus genu CCL2 nie miała wpływu na leczenie komórek TNF-α (rysunek 3F), co sugeruje, że transkrypcja nie zmieniła delecji pośredniczonej w CRISPR/Cas9., Aby jeszcze bardziej potwierdzić ten wynik, celowaliśmy w egzogenny Gen reporterski napędzany przez różne promotory o różnych mocach, gdzie różne działania transkrypcyjne mogły być oceniane za pomocą testu lucyferazy (rycina 3G). Testy PCR wykazały podobną skuteczność w celowanych delecjach w komórkach HEK 293T po Ko transfekcji genu reportera wraz z parami Cas9 i gRNA(ryc. 3h). Wynik ten wskazuje, że naprawa za pośrednictwem NHEJ może wystąpić pomimo występowania różnych stopni aktywności transkrypcyjnej.,
DSBs może stymulować HDR, aby umożliwić bardzo precyzyjne zastąpienie uszkodzonego regionu homologicznym dawcą. Aby uzyskać docelową genomową wymianę DNA, wprowadziliśmy parę grna, Cas9, i liniowy dawca z homologią do docelowego regionu w komórkach (rysunek 4A). Dawca liniowy otrzymywany był przez amplifikację PCR za pomocą primerów o sekwencji homologicznej 50 bp. Ten sam dawca został pomyślnie wstawiony za pomocą systemu naprawczego HDR opartego na ZFN (18)., Aby przetestować wykonalność zastąpienia CRISPR / Cas9 przez HDR, celowaliśmy w CCL2 locus z parą grna (#39 i #1854 pokazane na rysunku 3) i dawcą noszącym sekwencję kodowania enhanced Green fluorescent protein (EGFP) i miejsce SV40 poly(a) (rysunek 4A; sekwencje i pozycje są przedstawione w materiale uzupełniającym). Przy użyciu tego systemu około 0,5% komórek docelowych było EGFP-dodatnich, podczas gdy tylko 0,023% było EGFP-dodatnich w próbnych komórkach transfekcyjnych (po prostu transfekcyjnych z dawcą), co było podobne do komórek kontrolnych (0,021%, bez transfekcji)., Komórki EGFP-dodatnie zostały następnie posortowane za pomocą cytometrii przepływowej. Integracja Site-specific została potwierdzona przez PCR za pomocą dwóch par primerów flankujących zarówno ramiona homologiczne, jak i cały wymieniony region. Jak pokazano na rysunku 4B, zaobserwowaliśmy oczekiwany region zastępowany zawierający sekwencję EGFP o Pełnej długości i ramiona homologiczne (wynik sekwencjonowania Sangera pokazany w materiale uzupełniającym). Wykryto również endogenny allel typu dzikiego (ryc. 4B), co wskazuje, że nie wszystkie allele są celowane., Ponadto, wybraliśmy pojedyncze klony z komórek EGFP-dodatnich i okazało się, że wszystkie klony (6 z 6 badanych) miały oczekiwaną integrację( ryc. 4c), ale endogenny allel typu dzikiego został również wykryty w trzech klonach( ryc. 4C), co sugeruje, że tylko jeden allel był celowany w te klony. Ekspresja białka EGFP w komórkach celowanych (EGFP-positive sorted cells) była regulowana w górę w leczeniu TNF-α ocenianym przez Western blot i fluorescence activated cell sorting (FACS) (Rysunek 4, D i E)., Wyniki te wykazały, że system CRISPR/Cas9 może być używany do tworzenia zamienników genów / domen z wysoką wydajnością i dokładnością.

(A) Schematy przedstawiające procedurę ukierunkowanej wymiany genów za pomocą CRISPR / Cas9 w komórkach ludzkich., Aby sprawdzić skuteczność ukierunkowanej wymiany genów, guide RNAS (grnas) zaprojektowano tak, aby usunąć wskazany region (oznaczony przez miejsca 1 i 2) genu CCL2 i zastąpić usunięty region dawcą kasety EGFP-polyA z ramionami o krótkich regionach homologii. Docelowe miejsca w ośrodku 1 i Ośrodku 2 to #39 i #1854 w obrębie genu CCL2 pokazanego na rysunku 3. Sekwencje homologiczne (50 bp) znajdują się tuż przed i za miejscami delecji. Pozycje i sekwencje szczegółowo przedstawiono w materiale uzupełniającym., (B) skuteczność ukierunkowanej wymiany genu z CRISPR/Cas9 została określona przez PCR w komórkach HEK 293T. Do amplifikacji PCR wykorzystano podkłady łączące CCL2 i EGFP. C) test PCR dla docelowej wymiany genów pojedynczych klonów. (D,E) ekspresja białka EGFP po dodaniu TNF-α w komórkach HEK 293T została określona przez (D) Western blot i (E) FACS.
tutaj opisujemy proste i skuteczne podejście do delecji genów przy użyciu systemu CRISPR / Cas9., Pokazaliśmy, że wprowadzenie tego systemu do ludzkich komórek HEK 293t i innych ludzkich typów komórek, spowodowało delecje fragmentów do 10 kb z wydajnością w zakresie od 11% do 68%, w zależności od docelowej sekwencji. Możliwość skutecznego i precyzyjnego usuwania segmentów genomowych ułatwi badanie funkcjonalnych elementów genomowych w komórkach ludzkich. To podejście może potencjalnie używać celować jakaś genomic loci.
pojawiły się obawy dotyczące specyficzności systemu CRISPR/Cas9 (19-21)., Aby wykluczyć niechciane fenotypy z powodu mutacji poza celem, sugerujemy użycie co najmniej dwóch różnych par grna dla każdego regionu docelowego. W naszym badaniu potrzeba wielu par gRNA nie była głównym ograniczeniem, biorąc pod uwagę prostotę i wysoką wydajność tego systemu. Warto zauważyć, że różne pary grna skierowane do tego samego regionu działały z wysoką wydajnością (ryc. 2 i 4). Innym podejściem do unikania niechcianych mutacji jest zastosowanie metody podwójnego nickase (22, 23)., Z powodzeniem zastosowaliśmy również metodę double nickase do generowania delecji genomowego DNA, ale wydajność była znacznie niższa.
wiadomo, że naprawa DNA DSBs jest w dużej mierze pośredniczona przez podatny na błędy NHEJ, w którym oba końce są przetwarzane i ligowane razem w sposób, któremu często towarzyszą insercje i delecje nukleotydów. Takie podatne na błędy łączenie końcowe zaobserwowano w naprawie Dsb stworzonych przez Zfns lub TALENs. Natomiast naprawa Dsb generowanych przez Cas9 i dwa grna była bardzo precyzyjna., Nasze wyniki sugerują, że przerwy są bezpośrednio ligowane bez obróbki końcowej, ujawniając wcześniej niedocenianą zaletę szlaku NHEJ. Mechanizm, który powoduje dokładne ligacje pozostaje do ustalenia. Jedną z możliwości jest to, że ukierunkowana delecja za pomocą Cas9 i dwóch grna powoduje połączenie, które nie jest rozpoznawane przez żaden z oryginalnych grna. Analizowaliśmy również skuteczność w generowaniu mutacji indel dla poszczególnych par gRNA i gRNA (#39 i #224 gRNA na rysunku 3A) poprzez sekwencjonowanie Sangera AMPLIKONÓW PCR (klonowanie TA)., Co ciekawe, zaobserwowaliśmy, że wydajność generowania mutacji indel dla pojedynczego gRNA była dość niska (9,5%, 2 z 21 klonów dla #39 gRNA; 5%, 1 z 20 klonów dla #224 gRNA). Jednak para gRNA generowała wysoką skuteczność mutacji indel (50%, 10 z 20 klonów dla #39 i # 224), która była podobna do testu z użyciem qPCR (52%, rysunek 3B). Proponujemy, aby pojedynczy gRNA często skutkował jednym tępym końcem strony dekoltu, który zostanie precyzyjnie naprawiony przez NHEJ. Tak więc, efektywność generowania mutacji jest znacznie niższa przy użyciu pojedynczego gRNA niż pary grna.,
podziękowania
praca ta była wspierana przez granty Narodowego Instytutu Zdrowia (nr DP1CA174421) i W. M. Keck foundation do C.-Z. C oraz National Natural Science Foundation Of China (nr 81101481) i Shanghai Medical Talent Training Program (nr 81101481). XYQ2011048) do S. L. H. artykuł ten podlega Polityce publicznego dostępu NIH.
konkurencyjne interesy
autorzy nie deklarują konkurencyjnych interesów.
dane uzupełniające
aby zapoznać się z danymi uzupełniającymi dołączonymi do artykułu, odwiedź stronę internetową czasopisma: www.,future-science.com/doi/suppl/10.2144/000114196