Precis deleția genei și înlocuire folosind CRISPR/Cas9 sistem în celulele umane
Aici ne arată că doi ghid RNAs împreună cu Cas9 eficient de a genera ADN eliminări de până la 10 kb în celulele umane într-un proces în cazul în care repararea de stergere este în mare parte realizată către sfârșitul precis aderarea. În plus, furnizăm date care arată că sistemul CRISPR / Cas9 poate înlocui fragmente genomice mari în prezența unui donator de reparații liniare omoloage.,
repetițiile palindromice scurte, grupate regulat între bacterii/loci asociate cu CRISPR (CRISPR / Cas) codifică sistemele imunitare ghidate de ARN care protejează celulele împotriva virusurilor și plasmidelor invadatoare (1, 2). În Streptococcus pyogenes, sistemele CRISPR / Cas de tip II utilizează o endonuclează ghidată de ARN (rgen), Cas9, pentru a cataliza scindarea specifică site-ului secvențelor ADN țintă., Direcționare a Cas9 specifice genomice site-uri este mediată de un tânăr de 20 de nucleotide ghid secvență în cadrul unui asociat CRISPR ARN (crRNA) și necesită o trans-activarea crRNA (tracrRNA) care recrutează crRNA în Cas9 complex (3). Recunoașterea siturilor de clivaj este determinată de asocierea bazei ADN-CRRNA și de un motiv adiacent protospacerului (PAM), o secvență de trei nucleotide (NGG) juxtapusă la regiunea complementară ADN (4)., Este de remarcat că un singur ghid de ARN (gRNA) care imita tracrRNA-crRNA complexe pot recruta Cas9 pentru a vizat genomice site-uri și de a genera dublu catenar (DSBs) în ADN-ul (5). Sistemele CRISPR / Cas9 au fost adaptate pentru editarea genomului specific site-ului în diverse tipuri de celule și organisme (6-12).editarea genomului cu CRISPR / Cas9 este inițiată prin introducerea unui DSB la un locus genomic vizat folosind RGEN programat de ARN. Aceasta este urmată de repararea DSB fie prin repararea direcționată omologie (HDR) sau nonhomologous end-joining (NHEJ)., În prezența unui donator de reparații omolog, sistemul CRISPR/Cas9 poate fi utilizat pentru a genera modificări și inserții precise și definite la un locus vizat prin procesul HDR. În absența unui omolog de reparare donator, singur DSBs generate de CRISPR/Cas9 sunt reparate prin predispuse la erori NHEJ, ceea ce duce la inserarea sau ștergerea (indel) mutații. Mutațiile Indel în exonii de codificare pot introduce codoni de oprire prematură sau mutații de schimbare a cadrului, inactivând astfel proteinele corespunzătoare., Indel mutații generate de repararea unui singur DSB nu pot fi utile în experimente care vizează caracterizarea domeniilor funcționale de proteine-codare gene sau pentru inactivarea genomice elemente, cum ar fi intergenic sau intronic de reglementare secvențe sau noncoding ARN gene. Ștergerea fragmentelor de ADN din locația țintă ar oferi o cale de a studia aceste elemente funcționale. În acest scop, au fost introduse mai multe DSB-uri pentru a genera ștergeri în Drosophila (12, 13), zebrafish (14) și celule umane (8), deși cu eficiență scăzută., Vizate ADN genomic eliminări au fost, de asemenea, realizată folosind deget de zinc nuclează (ZFN) sau activator de transcriere-ca efectoare nuclează (TALEN) în celulele umane (15-17). Cu toate acestea, eficiența acestor abordări este în general scăzută. În plus, ZFNs și TALENs rămâne oarecum dificil și costisitor de a proiecta, dezvolta, și testăm în celulară context.aici, am examinat generarea de ștergeri de fragmente în celulele umane catalizate de sistemul CRISPR/Cas9. Arătăm că grn-urile 2 cuplate cu Cas9 pot crea eficient ștergeri de ADN de până la 10 kb., De interes, am constatat că repararea acestui proces de ștergere se realizează în mare măsură prin îmbinarea finală precisă. Mai mult, ștergerea vizată cu CRISPR/Cas9 pare să fie independentă de statutul transcripțional al locusului vizat. În cele din urmă, arătăm că sistemul CRISPR/ Cas9 poate fi utilizat pentru a înlocui fragmente genomice mari în prezența unui donator liniar de reparații omoloage.
Material și Metode
Plasmidă de construcție
De bază, H1 promotor a fost amplificat de pLVTHM plasmidic (Addgene, #12247, Cambridge, MA)., Oligonucleotidele care conțin promotorul H1 modificat și coloana vertebrală a secvențelor gRNA dorite cu două site-uri BsaI au fost sintetizate (PAN Facility, Universitatea Stanford). Produsele de lungime completă rezultate au fost amplificate prin PCR și clonate în vectorul pUC19. Ampicilina gene (amp) și H1 promotor în pUC19 vector conține BsaI enzimă de restricție site-uri; acestea au fost mutate (amp gena a fost schimbat de la G1601C, care nu modifică secvența de aminoacizi; H1 promotor a fost schimbat de la GAGACC să GAGGACC) pentru a elimina BsaI site-uri., Protocolul pentru clonarea gRNA este prezentat în materialul suplimentar. Toate secvențele site-urilor de direcționare sunt prezentate în tabelul suplimentar S1.
culturi de Celule
HEK 293T, SK-Hep1, și celulele HeLa au fost cultivate în Mediu de”s modified Eagle”s mediu (DMEM) suplimentat cu 10% ser fetal bovin (FBS) (Hyclone, Logan, UT) și penicilină/streptomicină (pen/streptococ) (Invitrogen, Carlsbad, CA). Celulele PC3 au fost cultivate în mediul RPMI-1640 suplimentat cu 10% FBS și pen/strep., Pentru factorul de necroză tumorală α (TNF-α) stimularea, 293T celulele au fost tratate cu concentrațiile indicate de TNF-α(R&D Sisteme, Minneapolis, MN). Celulele au fost menținute la 37°C și 5% CO2 într-un incubator umidificat.celulele HEK 293t au fost însămânțate în plăci cu 12 puțuri la o densitate de 100.000 de celule pe godeu. După 24 h, celulele au fost tranzitor transfectate cu 1 µg Cas9 plasmidic (Addgene, #41815), 0,5 µg gRNA T1, și 0,5 µg gRNA T2 plasmide folosind Lipofectamine 2000 (Invitrogen) ca pe producator”s-verbale., ADN-ul Genomic a fost extras la 48 de ore după transfecție folosind soluția de extracție a ADN-ului QuickExtract (Epicentre Biotechnologies, Madison, WI). PCR comun a fost realizat pentru a amplifica regiunea vizată folosind primeri care flancează regiunile vizate. Fragmentele genomice de tip sălbatic și trunchiate au fost rezolvate prin electroforeză în gel. PCR în timp Real (RT-PCR) a fost efectuat pentru a cuantifica procentul de deleție folosind primeri în joncțiune sau în regiunea de deleție. Metoda comparativă CQ a fost utilizată pentru a calcula nivelul de Expresie al regiunii țintă în raport cu o regiune de referință (Locus ACTB)., Procentul de deleție din regiunea țintă a fost calculat în continuare prin raportul dintre celulele țintă și celulele de control. Toate secvențele de grunduri sunt enumerate în tabelul suplimentar S2.
Țintă secvențiere
Celulele au fost recoltate două zile după transfecție, iar ADN-ul genomic a fost extras folosind QuickExtract Extracția ADN-ului Soluție (Epicentrul Biotehnologii). PCR a fost efectuat pentru a amplifica regiunea de direcționare cu ADN-ul genomic derivat din celule, iar ampliconii au fost secvențiați profund de sequencerul personal MiSeq (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Secvențele donatoare sunt prezentate în materialul suplimentar. Cele două 5′-cele mai multe legături sunt fosforotioat (indicat prin asteriscuri). Celulele din 6-plăci au fost tranzitor transfectate cu 2.0 µg Cas9 plasmidic, 0.8 µg gRNA T1 plasmidic, 0.8 µg gRNA T2 plasmidic, și 0,4 µg liniar donator folosind Lipofectamine 2000 (Invitrogen). La 48 de ore după transfecție, celulele au fost tratate cu 1 ng / mL TNF-α timp de 24 de ore, iar apoi celulele GFP pozitive au fost sortate.
Luciferază test
Pentru luciferază test, HEK 293T celulele au fost cultivate în plăci cu 96 de godeuri la o densitate de 5000 de celule pe ei., După 24 de ore, celulele au fost transfectate tranzitoriu cu 5 ng de reporter pRL-tk Renilla luciferase și 100 ng de reporter luciferase cu citomegalovirus (CMV), SV40 (virusul Simian 40) sau promotor de bază. După 48 de ore, activitatea luciferazei a fost măsurată cu sistemul dual de analiză a reporterului luciferazei (Promega).proteinele au fost separate prin pagina de dodecil sulfat de sodiu (SDS-PAGE) și transferate în membranele nitrocelulozei. Membranele au fost blocate cu 5% lapte degresat și incubate cu anticorpi GFP (CST, #2555S, Danvers, MA)., Complexul antigen-anticorp a fost detectat cu reactivi de chemiluminescență îmbunătățiți.
rezultate și discuții
am adaptat sistemul CRISPR/ Cas9 bacterian de tip II pentru a mutageniza ADN-ul genomic în celulele umane. Versiunea optimizată de codon uman a proteinei S. pyogenes Cas9 care poartă un semnal de localizare nucleară c-terminus SV40 a fost exprimată folosind un sistem descris anterior (6). Pentru a direcționa Cas9 clivaj la secvența dorită, ne-am exprimat crRNA-tracrRNA fuziune transcrieri, denumit în continuare ghid RNAs (gRNAs), la o modificare umane H1 polimeraza III promotor., Sfârșitul 3 ‘ al promotorului H1 a fost modificat pentru a permite transcrierea gRNAs care începe cu orice nucleotidă. Constrâns numai de cerința ca 20 bp crRNA țintă să fie urmată de PAM secvență, NGG (unde N este orice nucleotide), această abordare poate fi folosită în principiu pentru a viza orice genomice locație care are forma N20NGG. Pentru a facilita clonarea vectorului de Expresie gRNA, am folosit o enzimă de restricție de tip IIs, BsaI. Aceasta a necesitat sinteza unei oligonucleotide de 24 bp care conține o regiune de complementaritate cu situl țintă de pe ADN., Protocolul simplu și eficient pentru clonarea vectorului de Expresie gRNA (figura 1a) este descris în detaliu în materialul suplimentar.
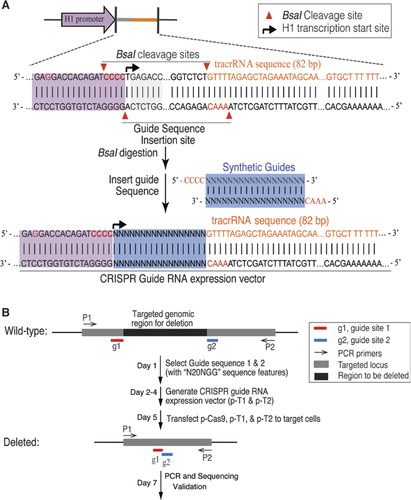
(A) proiectarea vectorului de Expresie ARN de ghidare (gRNA). Vectorul a fost proiectat pentru a produce gRNA transcrieri cu un sintetic gRNA topit la trans-activarea ARN/CRISPR ARN (tracrRNA)., H1 promotor a fost modificat pentru a elimina interne de tip IIS enzimă de restricție BsaI site-ului, prin schimbarea GAGACC în H1 promotor a GAGGACC. Un BsaI site-ul a fost introdus pentru a crea clonarea site-uri pentru gRNA și tracrRNA fuziuni de inserție a marcat sintetic duplexuri compatibil cu consolele. Capătul 3 ‘al promotorului H1 a fost modificat pentru a fi CCACAGATCCCC pentru a facilita transcrierea gRNAs cu orice nucleotidă la capătul 5’. (B) etapele de ștergere a genei vizate cu CRISPR/Cas9.,
Pentru eliminarea unui mare segment de ADN genomic, am folosit o pereche de gRNAs împotriva vizate locus (Figura 1B). Două locații țintă cu modelul N20NGG au fost selectate la limita regiunii țintă. Eficiența ștergerii direcționate ghidată de diverse combinații de perechi gRNA a fost determinată prin analize PCR folosind primeri care flancează regiunile vizate. Fragmentele genomice de tip sălbatic și trunchiate au fost rezolvate prin electroforeză în gel. Pentru a evita părtinirea amplificării PCR, eliminarea procentuală a fost cuantificată prin RT-PCR folosind o pereche de primeri., Primerii au fost proiectați în joncțiunea de ștergere (un primer în afara regiunii de ștergere, celălalt primer în regiunea de ștergere) sau în regiunile de ștergere (ambii primeri se află în regiunea de ștergere). Astfel, doar o singură bandă este amplificată cu perechea de grunduri atât pentru celulele vizate, cât și pentru celulele de control. Am calculat eliminarea procentuală prin compararea cantității relative de produse PCR (celule țintă versus celule de control) amplificate de aceeași pereche de primeri. Ștergerile vizate au fost verificate în continuare prin secvențiere.,
Pentru a evalua cât de gRNA perechi ar putea afecta ulterioare de reparații și generarea de eliminări, am proiectat prima seturi de gRNAs îndreptate împotriva omului CDC42 genomice locus și separate de distanțe variind de la aproximativ 200 la 10.000 bp (Figura 2A și Tabel Suplimentar S1). Apoi am evaluat capacitatea fiecărei perechi gRNA de a genera ștergeri în celulele umane HEK 293T în prezența Cas9. Eficiența robustă a ștergerilor bazate pe NHEJ (până la 68%) a fost confirmată de qPCR (figura 2b-2D)., Chiar și pentru ștergerea unei regiuni genomice de 10 kb, am obținut rate de direcționare de la 16% la 28%, în funcție de perechea gRNA. Acest proces de editare mediat de ARN a fost rapid, prima deleție detectabilă apărând la aproximativ 12 ore după transfecție (figura suplimentară S1). Sistemul a fost eficient într-o varietate de tipuri de celule, inclusiv: PC3, sk-Hep1 și celule HeLa (figura suplimentară S2).

(A) diagrama schematică care prezintă locațiile ARN-urilor de ghidare (gRNAs) care vizează locusul CDC42. (B–D) eficiența deleției țintite cu CRISPR/Cas9 a fost determinată prin PCR în celulele HEK 293t. S-au folosit primeri în afara regiunilor de ștergere preconizate. Procentul de deleție a fost cuantificat prin RT-PCR folosind primeri în joncțiune sau în regiunea de deleție. (E–f) eficiența și precizia ștergerii direcționate în CDC42 au fost confirmate prin (e) secvențiere Sanger și (F) analize de secvențiere cu randament ridicat. Produsul PCR care conține numai amplicon de ștergere a fost îmbogățit pentru secvențiere.,
Eliminări au fost ulterior confirmat prin secvențierea produșilor PCR întinde de așteptat clivaj site-uri. Secvențierea Sanger a arătat joncțiunile de ștergere rezultate din ligarea precisă a DSB-urilor cu capăt contondent create de Cas9; fiecare DSB a apărut exact cu 3 bp în amonte de secvența PAM (figura 2e și figura suplimentară S3). De asemenea, am folosit secvențierea profundă a ampliconilor de ștergere pentru a evalua acuratețea eficienței ștergerii; în aproximativ 80% din citiri, DSB-urile vizate au fost perfect reparate (figura 2f).,am recapitulat aceste constatări prin testarea perechilor gRNA concepute pentru a șterge fragmente dintr-un locus genomic care conține gena microRNA miR-21 din celulele HEK 293t. Două gRNAs au fost proiectate pentru a viza limitele acului mir-21 (figura suplimentară S4). Eficiența deleției a fost de 38% în urma transfecției cu cele două gRNAs și Cas9, măsurată utilizând un test PCR (figura suplimentară S4B). Secvențierea profundă a confirmat ștergerea a avut loc exact așa cum era de așteptat (figura suplimentară S4C).,
pentru a investiga dacă sistemul de ștergere a genei mediat de CRISPR/ Cas9 este influențat de starea transcripțională a genelor vizate, gena care codifică ligandul 2 de chemokină (C-C motif) (CCL2) a fost vizată. CCL2 este o citokină mică aparținând familiei de chemokine CC; gena CCL2 este o țintă a semnalizării NF-kB. Am selectat aleatoriu opt site-uri țintă situate în capetele 5′ și 3′ ale locusului genei CCL2 (figura 3a). Am realizat o ștergere robustă și eficientă a diferitelor regiuni ale genei folosind Cas9 și diferite perechi de gRNAs în celulele HEK 293T (figura 3b–3d).,

(A) diagrama schematică care prezintă locațiile ARN-urilor de ghidare (gRNAs) care vizează locusul CCL2. (B–D) eficiența eliminării direcționate a CCL2 cu CRISPR/Cas9 în celulele HEK 293t. (E) valorile ARNm CCL2 au fost determinate prin adăugarea factorului de necroză tumorală α (TNF-α) prin analize cantitative RT-PCR în celulele HEK 293t. Datele au fost prezentate cu mijloacele ± sem în trei exemplare., (F) eficiența eliminării direcționate a CCL2 cu CRISPR/Cas9 după tratamentul cu TNF-α Timp de 24 de ore în celulele HEK 293t. (G) nivelul activității luciferazei din citomegalovirus (CMV) sau SV40 sau un promotor de bază în celulele HEK 293t. Datele au fost prezentate cu mijloacele ± sem în trei exemplare. (H) eficiența deleției țintite a unei gene de luciferază controlată de CMV sau SV40 sau de un promotor de bază în celulele HEK 293t. (B-D, F, H) procentul de deleție a fost cuantificat prin RT-PCR folosind primeri în joncțiune sau în regiunea de deleție.,
Expresie a CCL2 gena a fost dramatic induse de TNF-α (până la ∼300 de ori creșterea) în HEK 293T celule (Figura 3E), oferind un bun model pentru a investiga dacă CRISPR/ Cas9 mediate vizat modificarea genomului este afectat de activitatea transcripțională. Interesant, eficiența deleției țintite a locusului genei CCL2 nu a fost afectată de tratamentul celulelor cu TNF-α(figura 3F), sugerând că transcripția nu a modificat deleția mediată de CRISPR/Cas9., Pentru a confirma în continuare acest rezultat, am vizat o genă reporter exogenă condusă de diverși promotori cu concentrații diferite, unde diferitele activități transcripționale ar putea fi evaluate folosind un test de luciferază (figura 3G). Testele PCR au evidențiat o eficiență similară în ștergerile direcționate în celulele HEK 293T după co-transfecția genei reporter împreună cu perechile Cas9 și gRNA (figura 3h). Acest rezultat indică faptul că reparația mediată de NHEJ poate apărea în ciuda apariției unor grade diferite de activitate transcripțională.,DSB-urile pot stimula HDR pentru a permite înlocuirea foarte precisă a regiunii deteriorate cu un donator omolog. Pentru a obține înlocuirea ADN-ului genomic vizat, am introdus o pereche de gRNAs, Cas9 și un donator liniar cu omologie pentru regiunea vizată în celule (figura 4a). Donatorul liniar a fost obținut prin amplificare PCR cu primeri care poartă o secvență omologă de 50 bp. Același donator a fost introdus cu succes folosind un sistem de reparare HDR bazat pe ZFN (18)., Pentru a testa fezabilitatea CRISPR/ Cas9 mediate de înlocuire de HDR, am vizat CCL2 locus cu o pereche de gRNAs (#39 și #1854 prezentată în Figura 3) și un donator poartă îmbunătățită proteina fluorescentă verde (EGFP) secvența de codificare și SV40 poli(O) ului (Figura 4A; secvențe și funcții sunt prezentate în Materialele Suplimentare). Folosind acest sistem, aproximativ 0,5% din celulele vizate au fost EGFP-pozitive, întrucât numai 0.023% au fost EGFP-pozitive în mock transfecția celulelor (doar transfectate cu donator), care a fost similar cu celulele control (0.021%, fără transfecție)., Celulele EGFP pozitive au fost apoi sortate prin citometrie în flux. Integrarea specifică sitului a fost confirmată prin PCR folosind două perechi de primeri care flancau atât brațele omoloage, cât și întreaga regiune înlocuită. După cum se arată în figura 4B, am observat Regiunea înlocuită așteptată care conține secvența EGFP de lungime completă și brațele omoloage (rezultatul secvențierii Sanger prezentat în materialul suplimentar). De asemenea, a fost detectată alela endogenă de tip sălbatic (figura 4B), indicând faptul că nu toate alelele sunt vizate., În plus, am selectat un singur clone din EGFP-pozitive celule și a constatat că toate clonele (6 din 6 examinat) a avut de așteptat de integrare (Figura 4C), dar endogene de tip sălbatic a fost, de asemenea, detectat în trei dintre clone (Figura 4C), sugerând că numai o alelă a fost vizat în acele clone. Expresie a EGFP proteine în celulele vizate (EGFP-pozitive sortate celule) a fost reglementată pe TNF-α tratament evaluată de către Western blot și fluorescență activated cell sorting (FACS) (Figura 4, D și E)., Aceste rezultate au demonstrat că sistemul CRISPR / Cas9 poate fi utilizat pentru a crea înlocuiri de gene/domenii cu eficiență și precizie ridicate.

(a) diagrame schematice care prezintă procedura de înlocuire a genelor vizate folosind CRISPR/Cas9 în celulele umane., Pentru a testa eficacitatea vizate genică de înlocuire, ghid RNAs (gRNAs) au fost concepute pentru a șterge indicat regiune (marcat de site-uri de 1 și 2) din CCL2 gene și să înlocuiască șters regiune cu EGFP-polyA casetă donator cu brațele scurte regiuni de omologie. Site-urile vizate ale site-ului 1 și site-ului 2 sunt #39 și #1854 în gena CCL2 prezentată în Figura 3. Secvențele omoloage (50 bp) sunt doar în amonte și în aval de site-urile de ștergere. Pozițiile și secvențele în detaliu sunt prezentate în materialul suplimentar., (B) eficiența înlocuirii genei țintite cu CRISPR/Cas9 a fost determinată prin PCR în celulele HEK 293t. Primerii care acoperă joncțiunile dintre CCL2 și EGFP au fost utilizați pentru amplificarea PCR. (C) testul PCR pentru înlocuirea genei țintă a clonelor unice. (D,E) expresia proteinei EGFP la adăugarea TNF-α în celulele HEK 293T a fost determinată de (D) Western blot și (E) FACS.
aici descriem o abordare simplă și eficientă pentru ștergerea genelor folosind sistemul CRISPR/Cas9., Am demonstrat că introducerea acestui sistem în om HEK 293T celule, și alte tipuri de celule umane, induse de eliminări de fragmente de până la 10 kb, cu randamente cuprinse între 11% și 68%, în funcție vizate secvență. Abilitatea de a șterge eficient și precis segmentele genomice va facilita studiul elementelor genomice funcționale din celulele umane. Această abordare poate fi utilizată potențial pentru a viza orice loci genomic.a existat îngrijorare cu privire la specificitatea sistemului CRISPR/Cas9 (19-21)., Pentru a exclude fenotipurile nedorite datorate mutațiilor în afara țintei, sugerăm să se utilizeze cel puțin două perechi diferite de gRNAs pentru fiecare regiune țintă. În studiul nostru, nevoia de mai multe perechi gRNA nu a fost o limitare majoră, având în vedere simplitatea și eficiența ridicată a acestui sistem. Este de remarcat faptul că diferite perechi de gRNAs orientate spre aceeași regiune au lucrat cu eficiență ridicată (figurile 2 și 4). O altă abordare pentru a evita mutațiile nedorite este utilizarea metodei duble nickase (22, 23)., De asemenea, am aplicat cu succes metoda dublă nickase pentru a genera ștergerea ADN-ului genomic, dar eficiența a fost considerabil mai mică.
este cunoscut faptul că de reparare a ADN-ului DSBs este mediată în mare parte de erori NHEJ, în care cele două capete sunt prelucrate și legate împreună într-un mod care este însoțită frecvent de nucleotide inserări și ștergeri. O astfel de eroare predispuse la sfârșitul aderarea a fost observată în repararea de DSBs create de ZFNs sau TALENs. În schimb, repararea DSB-urilor generate de Cas9 și două gRNAs a fost foarte precisă., Rezultatele noastre sugerează că pauzele sunt legate direct fără prelucrare finală, dezvăluind un avantaj anterior neapreciat al căii NHEJ. Mecanismul care are ca rezultat ligaturile precise rămâne de determinat. O posibilitate este că vizate stergere folosind Cas9 și două gRNAs rezultatele într-un nod care nu este recunoscut de către oricare dintre original gRNAs. De asemenea, am analizat eficiența în generarea indel mutații pentru individ gRNA și gRNA pereche (#39 și #224 gRNA în Figura 3A) de Sanger secvențiere a fragmentelor adn (TA clonare)., De interes, am observat că eficiența de generare a indel mutații pentru un singur gRNA a fost destul de redus (9.5%, 2 din 21 clone pentru #39 gRNA; 5%, 1 din 20 de clone pentru #224 gRNA). Cu toate acestea, perechea gRNA a generat o eficiență ridicată a mutațiilor indel (50%, 10 din 20 de clone pentru #39 și #224), care a fost similară cu testul folosind qPCR (52%, figura 3b). Noi propunem ca un singur gRNA de multe ori duce la un capăt bont al site-ului clivaj, care va fi reparat cu precizie de NHEJ. Astfel, eficiența generării mutației este mult mai mică cu utilizarea unui singur gRNA decât o pereche de gRNAs.,
mulțumiri
această lucrare a fost susținută prin granturi de la Institutul Național de Sănătate (nr. DP1CA174421) și W. M. Keck fundația C.-Z. C, și National Science Foundation Naturale din China (Nr 81101481) și Shanghai Medicale Talent Program de Formare (Nr. XYQ2011048) la S. L. H. această lucrare este supusă politicii de acces public NIH.
interese concurente
autorii nu declară interese concurente.
date suplimentare
pentru a vizualiza datele suplimentare care însoțesc această lucrare, vă rugăm să vizitați site-ul revistei la: www.,future-science.com/doi/suppl/10.2144/000114196