Exakt genborttagning och ersättning med hjälp av CRISPR / Cas9-systemet i mänskliga celler
här visar vi att två guide rna i kombination med Cas9 effektivt genererar DNA-deletioner på upp till 10 kb i mänskliga celler i en process där reparation av raderingen till stor del sker genom exakt slutfogning. Dessutom tillhandahåller vi data som visar att CRISPR / Cas9-systemet kan ersätta stora genomiska fragment i närvaro av en linjär homolog reparationsdonator.,
de bakteriella klustrade regelbundet interspaced korta palindromic repeats / CRISPR-associerade (CRISPR/Cas) loci kodar rna-styrda immunsystem som skyddar celler mot invaderande virus och plasmider (1, 2). I Streptococcus pyogenes använder typ II CRISPR/Cas-systemen ett RNA-styrt endonukleas (rgen), Cas9, för att katalysera platsspecifik klyvning av mål-DNA-sekvenser., Inriktning av Cas9 till specifika genomiska platser medieras av en 20 nukleotidstyrningssekvens inom ett associerat CRISPR RNA (crRNA) och kräver en transaktiverande crRNA (tracrna) som rekryterar crRNA till Cas9-komplexet (3). Erkännande av klyvningsställen bestäms av crRNA-DNA-basparning och ett protospacer-intilliggande motiv (PAM), en tre nukleotidsekvens (NGG) juxtaposed till DNA-komplementära regionen (4)., Det är anmärkningsvärt att en enda guide RNA (gRNA) som härmar tracrRNA-crRNA komplex kan rekrytera Cas9 riktade genomisk webbplatser och för att generera dubbelsträngat avbrott (DSBs) i DNA (5). CRISPR / Cas9-systemen har anpassats för områdesspecifik genomredigering i olika celltyper och organismer (6-12).
genomredigering med CRISPR / Cas9 initieras med införandet av en DSB på ett målinriktat genomiskt lokus med hjälp av RNA-programmerad RGEN. Detta är följt av reparation av DSB genom antingen homologi-regisserad reparation (HDR) eller nonhomologous end-anslutning (NHEJ)., I närvaro av en homolog reparationsdonator kan CRISPR / Cas9-systemet användas för att generera exakta och definierade modifieringar och Infogningar på ett målinriktat lokus genom HDR-processen. I avsaknad av en homolog reparationsdonator repareras enskilda Dsb som genereras av CRISPR / Cas9 genom den felbenägna nhej, vilket resulterar i införing eller radering (indel) mutationer. Indelmutationer i kodningsexoner kan införa för tidiga stoppkodon eller ramskiftmutationer och därigenom inaktivera motsvarande proteiner., Indelmutationer som genereras från att reparera en enda DSB kanske inte är användbara i experiment som syftar till att karakterisera de funktionella domänerna för proteinkodande gener eller för inaktivering av genomiska element såsom intergena eller introniska reglerande sekvenser eller icke-kodande RNA-gener. DNA fragment deletioner i mål loci skulle ge en väg att studera dessa funktionella element. För detta ändamål har flera Dsb införts för att generera deletioner i Drosophila (12, 13), zebrafisk (14) och mänskliga celler (8), Om än med låg effektivitet., Riktade genomiska DNA-deletioner har också uppnåtts med användning av zinkfingerkärnor (ZFN) eller transkriptionsaktivatorliknande effektorkärnor (talan) i humana celler (15-17). Effektiviteten i dessa tillvägagångssätt är dock i allmänhet låg. Dessutom förblir ZFNs och TALENs något svåra och dyra att designa, utveckla och empiriskt testa i mobilkontexten.
här undersökte vi generering av fragmentraderingar i mänskliga celler som katalyseras av CRISPR/Cas9-systemet. Vi visar att 2 gRNAs tillsammans med Cas9 effektivt kan skapa DNA strykningar av upp till 10 kb., Av intresse fann vi att reparation av denna borttagningsprocess till stor del uppnås genom exakt slutfogning. Dessutom verkar målinriktad radering med CRISPR/Cas9 vara oberoende av den transkriptionella statusen för det riktade loket. Slutligen visar vi att CRISPR / Cas9-systemet kan användas för att ersätta stora genomiska fragment i närvaro av en linjär homolog reparationsdonator.
Material och metoder
Plasmidkonstruktion
den grundläggande H1-promotorn förstärktes från pLVTHM plasmid (Addgene, #12247, Cambridge, MA)., Oligonukleotider som innehåller modifierade H1 projektansvarig och ryggraden i önskad gRNA sekvenser med två BsaI platser syntetiseras (PAN Anläggning, Stanford University). De resulterande fullängdsprodukterna förstärktes av PCR och klonades i pUC19-vektorn. Den ampicillin gen (amp) och H1 promotor i pUC19 vektor innehåller BsaI begränsning enzym platser, dessa var muterade (amp gen har ändrats från G1601C, som inte förändrar den aminosyrasekvens; H1 arrangören har ändrats från GAGACC att GAGGACC) för att eliminera BsaI webbplatser., Protokollet för kloning av gRNA presenteras i Tilläggsmaterialet. Alla inriktningsplatser sekvenser presenteras i Tilläggstabell S1.
Cell kultur
HEK 293T, SK-Hep1, och HeLa celler odlades i Dulbecco”s modified Eagle”s medium (DMEM) kompletteras med 10% fetalt bovint serum (FBS) (Hyclone, Logan, UT) och penicillin/streptomycin (pen/strep) (Invitrogen, Carlsbad, CA). PC3 cellerna odlades i RPMI-1640 medium kompletteras med 10% FBS och penna/strep., För stimulering av tumörnekrosfaktor α (TNF-α) behandlades 293T-celler med angivna koncentrationer av TNF-α (R&d-system, Minneapolis, MN). Cellerna bibehölls vid 37 ° C och 5% CO2 i en fuktad inkubator.
riktad DNA-radering
HEK 293T-celler frös i 12-brunnsplattor med en densitet av 100 000 celler per brunn. Efter 24 h, cellerna var kortvarigt transfected med 1 µg Cas9 plasmid (Addgene, #41815), 0.5 µg gRNA T1 och 0,5 µg gRNA T2 plasmider med Lipofectamine 2000 (Invitrogen) enligt tillverkarens protokoll., Genomiskt DNA extraherades 48 h efter transfection med QuickExtract DNA-Extraktion Lösning (Epicentrum Bioteknik, Madison, WI). Gemensam PCR genomfördes för att förstärka den riktade regionen med hjälp av primers flankerar de riktade regionerna. Vildtyp och stympade genomiska fragment löstes genom gelelektrofores. Real-time PCR (RT-PCR) utfördes för att kvantifiera procent av raderingen med hjälp av primers över korsningen eller inom raderingsområdet. Den jämförande Cq-metoden användes för att beräkna uttrycksnivån för målregionen i förhållande till en referensregion (ACTB locus)., Procent av borttagningen i målregionen beräknades ytterligare av förhållandet mellan målceller i förhållande till kontrollceller. Alla primersekvenser listas i Tilläggstabell S2.
Target sequencing
celler skördades två dagar efter transfektion, och det genomiska DNA extraherades med hjälp av QuickExtract DNA Extraction Solution (Epicentre Biotechnologies). PCR genomfördes för att förstärka målområdet med genomiskt DNA härrörande från cellerna, och amplicons var djupt sekvenserade av MiSeq Personal Sequencer (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., Givarsekvenserna presenteras i det kompletterande materialet. De två 5′-de flesta länkarna är fosforotioat (indikeras av asterisker). Celler i 6-brunnsplattor transfekterades transient med 2.0 µg Cas9 plasmid, 0.8 µg gRNA T1 plasmid, 0.8 µg gRNA T2 plasmid och 0.4 µg linjär givare med Lipofektamin 2000 (Invitrogen). Vid 48 h efter transfektion behandlades celler med 1 ng/mL TNF-α för 24 h och sedan sorterades GFP-positiva celler.
Luciferaseanalys
för luciferaseanalysen såddes HEK 293T-celler i 96-brunnsplattor med en densitet av 5000 celler per brunn., Efter 24 h, cellerna var kortvarigt transfected med 5 ng av pRL-TK Renilla luciferas reporter och 100 ng luciferas reporter med cytomegalovirus (CMV), SV40 (Simian virus 40), eller grundläggande projektansvarig. Efter 48 timmar mättes luciferasaktiviteten med dual luciferase reporter assay system (Promega).
Western blot
proteiner separerades av natriumdodecylsulfat—sida (SDS-sida) och överfördes till nitrocellulosmembran. Membranen blockerades med 5% fettfri mjölk och inkuberades med GFP antikropp (CST, #2555S, Danvers, MA)., Antigenantikroppskomplexet detekterades med förbättrade kemiluminescensreagenser.
resultat och diskussion
vi anpassade det bakteriella typ II CRISPR / Cas9-systemet för att mutagenisera genomiskt DNA i mänskliga celler. Den mänskliga kodonoptimerade versionen av S. pyogenes Cas9-protein som bär en C-terminus SV40-nukleär lokaliseringssignal uttrycktes med hjälp av ett tidigare beskrivet system (6). Att direkt Cas9 klyvning till önskad sekvens, vi uttryckte crRNA-tracrRNA fusion avskrifter, nedan kallad guide Rna (gRNAs), från en modifierad mänskliga H1-polymeras III projektansvarig., 3-änden av H1-promotorn ändrades för att tillåta transkription av gRNAs som börjar med någon nukleotid. Begränsas endast av kravet att 20 bp crRNA-målet ska följas av PAM-sekvensen, NGG (där N är någon nukleotid), kan detta tillvägagångssätt i princip användas för att rikta sig mot vilken genomisk plats som helst som har formen N20NGG. För att underlätta kloning av grna expression vector använde vi ett typ IIS restriktionsenzym, BsaI. Detta krävde syntesen av en 24 bp-oligonukleotid innehållande en komplementaritetsregion till målplatsen på DNA., Det enkla och effektiva protokollet för kloning av grna-uttrycksvektorn (Figur 1A) beskrivs i detalj i det kompletterande materialet.
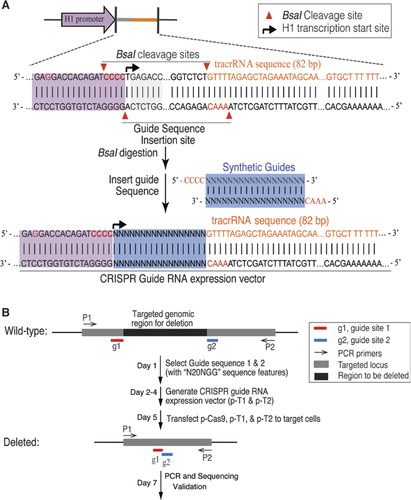
(a) utformning av styr-rna-expressionsvektorn (grna). Vektorn var utformad för att producera grna-transkript med en syntetisk gRNA smält till det transaktiverande RNA / CRISPR RNA (tracrRNA)., H1 initiativtagare var modifierad för att eliminera den interna typ IIS begränsning enzym BsaI webbplats genom att ändra GAGACC i H1 projektansvarig GAGGACC. En bsai plats infördes för att skapa kloning platser för gRNA och tracrRNA fusioner genom införande av syntetiska oligonukleotid duplexer med kompatibla överhäng. 3 ”i slutet av H1 initiativtagare var modifierad för att vara CCACAGATCCCC för att underlätta transkriberingen av gRNAs med någon nukleotid på 5′ änden. B) stegen för riktad genborttagning med CRISPR/Cas9.,
för radering av ett stort segment av genomiskt DNA använde vi ett par gRNAs mot riktade lokus (Figur 1b). Två målplatser med mönstret N20NGG valdes ut vid gränsen för målregionen. Effektiviteten av målinriktad radering styrd av olika kombinationer av gRNA-par bestämdes genom PCR-analyser med hjälp av primers som flankerar de riktade regionerna. Vildtyp och stympade genomiska fragment löstes genom gelelektrofores. För att undvika PCR amplifiering bias, procent radering kvantifierades av RT-PCR med hjälp av en primer par., Primers designades över radering korsningen (en primer utanför radering regionen, den andra primer inom radering regionen) eller inom radering regioner (båda primers lokalisera inom radering regionen). Således förstärks endast ett enda band med primerparet för både riktade celler och kontrollceller. Vi beräknade procentuell radering genom att jämföra den relativa mängden PCR-produkter (målceller mot kontrollceller) förstärkta av samma primerpar. Riktade raderingar verifierades ytterligare genom sekvensering.,
för Att bedöma hur gRNA par kan påverka efterföljande reparation och generering av strykningar, vi första utformad uppsättningar av gRNAs riktade mot den mänskliga CDC42 genetiska lokus och separerad genom avstånd som sträcker sig från cirka 200 till 10 000 bp (Figur 2A och Kompletterande Tabell S1). Vi bedömde sedan förmågan hos varje gRNA-par att generera deletioner i mänskliga HEK 293T-celler i närvaro av Cas9. Robusta effektivitetsvinster av nhej–baserade deletioner (upp till 68%) bekräftades av qPCR (Figur 2B-2D)., Även för radering av en 10 kb genomisk region fick vi inriktningshastigheter på 16% till 28%, beroende på gRNA-paret. Denna rna-medierade redigeringsprocess var snabb, med den första detekterbara raderingen som uppträdde ungefär 12 h efter transfektion (kompletterande figur S1). Systemet var effektivt i en mängd olika celltyper, inklusive: PC3, SK-Hep1 och HeLa-celler (kompletterande figur S2).

(a) schematiskt diagram som visar platserna för guide RNAs (gRNAs) som riktar sig till Cdc42-lokusen. (B–D) effektiviteten av riktade radering med CRISPR/Cas9 bestäms av PCR i HEK 293T celler. Primers utanför förväntade deletionsregioner användes. Procentandelen av raderingen kvantifierades av RT-PCR med hjälp av primers över korsningen eller inom raderingsområdet. (E–F) effektiviteten och precisionen av riktad radering i CDC42 bekräftades genom (e) Sanger sekvensering och (F) hög genomströmning sekvensanalyser. PCR-produkten som endast innehåller deletion amplicon berikades för sekvensering.,
deletioner bekräftades ytterligare genom sekvensering av PCR-produkter som spänner över de förväntade klyvningsställena. Sanger-sekvensering visade radering korsningar resulterade i exakt ligering av den trubbiga-slutade DSBs skapats av Cas9; varje DSB inträffade exakt 3 bp uppströms av PAM-sekvens (Figur 2E och Kompletterande Figur S3). Vi använde också djup sekvensering av radering amplicons för att bedöma riktigheten av radering effektivitet; i ca 80% av läser, riktade DSBs var perfekt reparerade (figur 2F).,
vi rekapitulerade dessa fynd genom att testa gRNA-par som är utformade för att ta bort fragment från ett genomiskt lokus som innehåller mirorna miR-21-genen i HEK 293T-celler. Två gRNAs var utformade för att rikta gränser miR-21 hårnål (Kompletterande Figur S4). Radering effektivitet var 38% följande transfection med två gRNAs och Cas9, mätt med hjälp av en PCR-analys (Kompletterande Figur S4B). Djup sekvensering bekräftade raderingen inträffade exakt som förväntat (kompletterande figur S4C).,
för att undersöka om CRISPR/ Cas9-medierade raderingssystemet påverkas av transkriptionstillståndet hos riktade gener, var genen som kodade chemokine (c-c motif) ligand 2 (CCL2) riktad. CCL2 är en liten cytokin som tillhör CC chemokine familj; CCL2 gen är ett mål för NF-kB-signalering. Vi valde slumpmässigt åtta målplatser i 5′ och 3 ’ ändarna av CCL2 gen locus (figur 3a). Vi uppnått robust och effektiv borttagning av olika regioner i gen med hjälp av Cas9 och olika par av gRNAs i HEK 293T celler (Figur 3B–3D).,

(a) schematiskt diagram som visar platserna för guide RNAs (gRNAs) som riktar sig till CCL2-lokusen. (B–D) effektiviteten av riktade radering av CCL2 med CRISPR/Cas9 i HEK 293T celler. E) CKL2 mRNA-nivåer bestämdes vid tumörnekrosfaktor α (TNF-α) tillsats genom kvantitativa RT-PCR-analyser i HEK 293T-celler. Data visades med medelvärdet ± sem i tre exemplar., (F) effektiviteten av riktade radering av CCL2 med CRISPR/Cas9 efter behandling med TNF-α för 24 h i HEK 293T celler. (G) Den nivå av luciferas aktivitet från cytomegalovirus (CMV), eller SV40, eller en grundläggande projektansvarig i HEK 293T celler. Data visades med medelvärdet ± sem i tre exemplar. H) effektivitet vid riktad radering av en luciferasgen som kontrolleras av CMV, eller SV40, eller en grundläggande promotor i HEK 293T-celler. (B–D,F,H) procentandelen av raderingen kvantifierades av RT-PCR med hjälp av primrar över korsningen eller inom raderingsområdet.,
Uttryck för CCL2 gen var dramatiskt inducerad av TNF-α (upp till ∼300-faldig ökning) i HEK 293T celler (Figur 3E), som ger en bra modell för att undersöka om CRISPR/ Cas9-medierad riktade genomet redigering påverkas av transkriptionell aktivitet. Av intresse påverkades inte effektiviteten av riktad radering av CCL2 – genen locus av behandling av celler med TNF-α(figur 3F), vilket tyder på att transkription inte förändrade CRISPR/Cas9-medierad radering., För att ytterligare bekräfta detta resultat riktade vi oss till en exogen reporter-gen som drivs av olika promotorer med olika styrkor, där de olika transkriptionsaktiviteterna kunde utvärderas med hjälp av en luciferase-analys (figur 3G). PCR-analyser visade liknande effektivitet i riktade strykningar i HEK 293T celler efter co-transfection av reportern gen tillsammans med Cas9 och gRNA par (Figur 3H). Detta resultat indikerar att nhej-medierad reparation kan inträffa trots förekomsten av varierande grad av transkriptionell aktivitet.,
Dsb kan stimulera HDR för att möjliggöra mycket exakt ersättning av den skadade regionen med en homolog givare. För att erhålla riktad genomisk DNA-ersättning introducerade vi ett par gRNAs, Cas9 och en linjär givare med homologi till den riktade regionen i celler (figur 4A). Den linjära givaren erhölls genom PCR-förstärkning med primrar som bär en 50 bp av homolog sekvens. Samma givare infördes framgångsrikt med ett ZFN-baserat HDR-reparationssystem (18)., För att undersöka genomförbarheten av CRISPR/ Cas9-medierad ersättning med HDR, vi riktade CCL2 locus med ett par gRNAs (#39 och #1854 visas i Figur 3) och en givare med förbättrad grönt fluorescerande protein (EGFP) kodande sekvens och SV40 poly(A) webbplats (Figur 4A, sekvenser och positioner redovisas i den Kompletterande Material). Med hjälp av detta system var cirka 0,5% av riktade celler EGFP-positiva, medan endast 0,023% var EGFP-positiva i mock-transfektionsceller (bara transfekterade med givare), vilket liknade kontrollceller (0,021%, utan transfektion)., De EGFP-positiva cellerna sorterades sedan efter flödescytometri. Områdesspecifik integration bekräftades av PCR med hjälp av två par primrar som flankerar både homologa armar och hela den ersatta regionen. Som visas i Figur 4B observerade vi den förväntade ersatta regionen som innehåller EGFP-sekvensen med full längd och de homologa armarna (Sanger-sekvenseringsresultatet visas i Tilläggsmaterialet). Den endogena vildtypsallelen upptäcktes också (figur 4B), vilket indikerar att inte alla alleler är riktade., Dessutom valde vi enstaka kloner från EGFP-positiva celler och fann att alla kloner (6 av 6 undersökta) hade den förväntade integrationen (figur 4C), men den endogena vildtypsallelen upptäcktes också i tre av klonerna (figur 4C), vilket tyder på att endast en allel var riktad i dessa kloner. Uttryck av EGFP-protein i riktade celler (EGFP-positiva sorterade celler) var uppreglerat på TNF-α-behandling som utvärderats av Western blot och fluorescens aktiverad cellsortering (FACS) (Figur 4, D och E)., Dessa resultat visade att CRISPR/Cas9-systemet kan användas för att skapa gen/domänersättningar med hög effektivitet och noggrannhet.

(A) schematiska diagram som visar förfarandet för riktad genersättning med CRISPR/Cas9 i humana celler., För att testa effekten av riktad genersättning, guide RNAs (gRNAs) utformades för att ta bort den angivna regionen (markerad av platserna 1 och 2) av CCL2-genen och att ersätta den borttagna regionen med EGFP-polyA kassettdonatorn med armar med korta regioner av homologi. De riktade platserna på plats 1 och plats 2 är # 39 och # 1854 inom CCL2-genen som visas i Figur 3. De homologa sekvenserna (50 bp) är bara uppströms och nedströms borttagningsplatserna. Positionerna och sekvenserna i detalj presenteras i det kompletterande materialet., (B) effektiviteten av riktade gen ersättning med CRISPR/Cas9 bestäms av PCR i HEK 293T celler. Primers som spänner över korsningar mellan CCL2 och EGFP användes för PCR-amplifiering. C) PCR-analys för riktad genersättning av de enskilda klonerna. (D,E) Uttryck för EGFP protein på TNF-α förutom i HEK 293T celler bestäms av (D) Western blot och (E) FACS.
här beskriver vi ett enkelt och effektivt tillvägagångssätt för genborttagning med CRISPR / Cas9-systemet., Vi visade att införandet av detta system i mänskliga HEK 293T-celler, och andra mänskliga celltyper, inducerade deletioner av fragment upp till 10 kb med effektivitetsvinster mellan 11% och 68%, beroende på den riktade sekvensen. Förmågan att effektivt och exakt radera genomiska segment kommer att underlätta studien av funktionella genomiska element i mänskliga celler. Detta tillvägagångssätt kan potentiellt användas för att rikta alla genomiska loci.
det finns oro för CRISPR/Cas9-systemets specificitet (19-21)., För att utesluta oönskade fenotyper på grund av off-target-mutationer föreslår vi att minst två olika par gRNAs används för varje målregion. I vår studie var behovet av flera grna-par inte en stor begränsning, med tanke på enkelheten och effektiviteten i detta system. Det är anmärkningsvärt att olika par gRNAs riktade till samma region arbetade med hög effektivitet (figurerna 2 och 4). Ett annat tillvägagångssätt för att undvika oönskade mutationer är användningen av dubbel nickase-metoden (22, 23)., Vi tillämpade också framgångsrikt den dubbla nickasmetoden för att generera radering av genomiskt DNA, men effektiviteten var betydligt lägre.
det är känt att reparationen av DNA-Dsb är till stor del medierad av felbenägen NHEJ, där de två ändarna bearbetas och ligeras tillsammans på ett sätt som ofta åtföljs av nukleotid Infogningar och deletioner. Sådan felbenägen slutförbindelse observerades vid reparation av DSBs som skapats av ZFNs eller TALENs. Däremot var reparationen av DSB som genererades av Cas9 och två gRNAs mycket exakt., Våra resultat tyder på att rasterna är direkt ligerade utan slutbehandling, vilket avslöjar en tidigare ouppskattad fördel med nhej-vägen. Den mekanism som resulterar i de exakta ligationerna återstår att bestämma. En möjlighet är att riktad radering med hjälp av Cas9 och två gRNAs resulterar i en korsning som inte känns igen av någon av de ursprungliga gRNAs. Vi analyserade också effektiviteten i att generera indelmutationer för det enskilda grna-och grna-paret (#39 och # 224 gRNA i figur 3A) genom Sanger-sekvensering av PCR-ampliconer (ta-kloning)., Av intresse observerade vi att effektiviteten att generera indelmutationer för enstaka gRNA var ganska låg (9.5%, 2 av 21 kloner för #39 gRNA; 5%, 1 av 20 kloner för #224 gRNA). Grna-paret genererade emellertid hög effektivitet av indelmutationer (50%, 10 av 20 kloner för #39 och #224), vilket liknade analysen med qPCR (52%, figur 3b). Vi föreslår att en enda gRNA ofta resulterar i en trubbig ände av klyvningsplatsen, som kommer att repareras exakt av NHEJ. Således är effektiviteten att generera mutation mycket lägre med användning av en enda gRNA än ett par grna.,
bekräftelser
detta arbete stöddes av bidrag från National Institute of Health (nr. DP1CA174421) och W. M., Keck foundation-c-Z. C, och National Natural Science Foundation of China (Nr 81101481) och Shanghai Medical Talent Training Program (Nr. XYQ2011048) att S. L. H. Detta dokument är föremål för NIH Allmänhetens Tillgång Politik.
konkurrerande intressen
författarna förklarar inga konkurrerande intressen.
kompletterande uppgifter
för att se de kompletterande uppgifter som åtföljer detta dokument, besök tidskriftens webbplats på: www.,future-science.com/doi/suppl/10.2144/000114196