Jaké jsou rozdíly ve třídách zdravotnických prostředků FDA?
US FDA reguluje všechny zdravotnické prostředky uváděné na trh v USA, které jsou seskupeny do tří širokých tříd. Jakýkoli zdravotnický prostředek schválený FDA je klasifikován jako buď třídy I, II, nebo III v závislosti na riziku zařízení, invazivita, a dopad na celkový zdravotní stav pacienta. Ale kde jsou čáry nakreslené mezi každou z těchto tří tříd a proč?,
pokyny pro klasifikaci US FDA mohou být velmi matoucí pro výrobce zdravotnických prostředků s omezenou expozicí systému. Existuje obrovský rozdíl v optimální cestě k trhu pro výrobce v závislosti na tom, jak je vaše zařízení seskupeno. Zařízení třídy I podléhají mnohem menším regulačním požadavkům než zařízení třídy II nebo III.
pochopením rozdílů ve třídách zdravotnických prostředků FDA můžete pochopit, jak bude vaše zařízení seskupeno., S těmito znalostmi v ruce mohou výrobci zdravotnických prostředků v etapách premarketu lépe připravit a přidělit zdroje potřebné pro regulační schválení.
rozdíly mezi třídami zdravotnických prostředků FDA
FDA klasifikovala více než 1700 různých typů zdravotnických prostředků. Zařízení jsou organizována v Kodexu federálních předpisů (CFR) podle 16 specialit, jako jsou kardiovaskulární nebo Hematologická zařízení., Klasifikace zdravotnického prostředku podle jedné z 16 specialit je prvním krokem k pochopení toho, zda vyrábíte zdravotnické zařízení třídy I, II nebo III.
Po zařazení zařízení podle specializace, FDA pověřuje výrobci přistoupit k premarket oznámení se znalostí, zda je jejich přístroj je osvobozeno od daně nebo není. Zdravotnické prostředky třídy I, nejméně riskantní a invazivní kategorie, jsou osvobozeny od procesů oznámení premarketu. Zvláštní zařízení třídy II jsou rovněž osvobozena od schválení premarketu.,
všechna zařízení regulovaná FDA však podléhají současným požadavkům na správnou výrobní praxi (cGMP) pro registraci, označování a kvalitu. Ale jak víte, zda je vaše zařízení třídy I nebo II, a zda jste povinni podstoupit oznámení premarket? 
1. Třídy
FDA definuje Třídy I zařízení jako zařízení, které „nejsou určeny pro použití v podporu nebo udržení života nebo zásadní význam v prevenci poškození pro lidské zdraví, a nesmí představovat nepřiměřené riziko onemocnění nebo zranění.,“
tato zařízení jsou nejběžnější třídou zařízení regulovaných FDA, což představuje 47 procent schválených zařízení na trhu.
zařízení třídy I mají minimální kontakt s pacienty a nízký dopad na celkové zdraví pacienta. Zařízení třídy I obecně nepřicházejí do styku s vnitřními orgány pacienta, centrálním nervovým systémem nebo kardiovaskulárním systémem. Tato zařízení podléhají nejmenším regulačním požadavkům.,
Příklady Třída Zařízení:
- Kartáček
- Špachtle
- Kyslíková Maska
- opakované použití Chirurgického Skalpelu
- Obvazy
- Nemocniční Lůžka
Přináší. Třídy Zdravotnických Prostředků na Trh
Třídy I zařízení jsou nejrychlejší a nejjednodušší uvést na trh, protože představují nejnižší míru rizika pro pacienta, a jen zřídka jsou rozhodující pro život-udržení péče. Většina zařízení třídy I je osvobozena od požadavků FDA na oznámení Premarketu (510k) a schválení Premarketu (PMA).,
zařízení třídy I nejsou osvobozena od obecných kontrol FDA, což je řada příkazů, které se vztahují na zdravotnické prostředky třídy I, II a III. Ustanovení tohoto zákona se zabývají cizoložstvím, nesprávným označením, registrací zařízení, záznamy a dobrými výrobními postupy. Výrobci zdravotnických prostředků, kteří spadají do třídy a, jsou stále povinni implementovat systém řízení kvality a dodržovat standardy pro zajištění kvalitního produktu.,
SOUVISEJÍCÍ ČTENÍ: Rozdíl Mezi Premarket Notification 510(k) a Premarket Schválení
2. Třídy
Třídy II zdravotnických zařízení jsou složitější než u prostředků Třídy I a představují vyšší kategorii rizika, protože oni jsou více pravděpodobné, že přijde do trvalého kontaktu s pacientem. To může zahrnovat zařízení, která přicházejí do styku s kardiovaskulárním systémem pacienta nebo vnitřních orgánů, a diagnostické nástroje.,
FDA definuje zařízení třídy II jako “ zařízení, pro která obecné kontroly nestačí k zajištění přiměřené jistoty bezpečnosti a účinnosti zařízení.,est Soupravy
Uvedení Třídy II Zdravotnických Prostředků na Trh
Ovládání se liší v závislosti na zařízení, ale podle FDA, může zahrnovat:
- výkon Zařízení
- Postmarket sledování
- registry Pacientů
- Speciální požadavky na označování
- Premarket požadavky na data
- Doporučení
většina z II. Třídy zařízení FDA schválených pro trh prostřednictvím Premarket Oznámení, nebo 510(k) procesu.,
zařízení třídy II podléhají stejným obecným kontrolám uvedeným výše, ale FDA je definuje jako “ zařízení, pro která obecné kontroly nestačí k zajištění přiměřené jistoty bezpečnosti a účinnosti zařízení.“Z tohoto důvodu jsou zařízení třídy II také předmětem zvláštních ovládacích prvků. Tyto předpisy závisí na zařízení a mohou zahrnovat zvláštní požadavky na označování, registry pacientů a standardy výkonu.
většina zařízení třídy II přichází na trh pomocí procesu oznámení Premarket (510K)., 510 (k) je komplexní aplikace pro FDA, která ukazuje, že zařízení je bezpečné a účinné tím, že prokazuje, že zařízení je ekvivalentní jinému zařízení, které je na trhu.
tento proces zahrnuje zobrazení “ podstatné rovnocennosti „s jiným zařízením, které je v jazyce FDA známé jako“ predikát.“To neznamená, že zařízení musí být identické, ale vyžadují značné podobnosti v použití, design, materiály, označování, normy, a další vlastnosti.,
FDA vydala seznam výjimek na začátku roku 2018, který osvobozuje více než 800 generických zdravotnických prostředků třídy I a II od procesu 510(k). Pokud máte obecný zdravotnický prostředek třídy II, můžete zjistit, zda je osvobozen od podání 510(k) hledáním databáze klasifikace produktů FDA.
související čtení: 5 důvodů Generální opravy FDA 510 (k) je skvělý krok.
Třída 3
FDA definuje zařízení třídy III jako produkty, které “ obvykle udržují nebo podporují život, jsou implantovány nebo představují potenciální nepřiměřené riziko onemocnění nebo zranění.,“
Jen 10 procent zařízení, řídí NÁS FDA spadají do Třídy III. Tato klasifikace je všeobecně rozšířena na trvalé implantáty, inteligentní lékařské zařízení a systémy podpory života.
zatímco třída III je obecně vyhrazena pro nejinovativnější a nejmodernější zdravotnické prostředky, existují další zařízení, která mohou z různých důvodů spadat do třídy III., Některé přístroje, které jsou rozděleny do kategorií zpočátku jako Třída II, může být povýšen do Třídy III, pokud výrobce není schopen prokázat podstatné rovnocennosti k predikátu (stávající produkt) během PMA (510k) podání procesu.,
Příklady III. Třída Zdravotnické Zařízení:
- Prsní implantáty
- Kardiostimulátory
- Defibrilátory
- vysokofrekvenční ventilátory
- Kochleární implantáty
- Fetální krve odběr vzorků monitory
- Implantované protézy
Uvedení Třídy III Zdravotnických Prostředků na Trh
Prostředky třídy III se vztahuje na všechny FDA Obecné Ovládací prvky a FDA Premarket Schválení (PMA) proces., FDA píše: „vzhledem k úrovni rizika spojeného se zařízeními třídy III FDA zjistila, že samotné obecné a speciální kontroly nejsou dostatečné k zajištění bezpečnosti a účinnosti zařízení třídy III.“
PMA je nejintenzivnější typ aplikace pro marketing zařízení vyžadované FDA. Některá zařízení FDA třídy III jsou osvobozeny a mohou mít nárok na 510 (k) podání, ale většina se očekává, že získat schválení Premarket.,
proces PMA vyžaduje důslednou studii zdravotnického prostředku, aby prokázal bezpečnost a účinnost prostřednictvím vývoje profilu přínosu/rizika založeného na datech. Proces PMA obecně zahrnuje klinické studie a významný čas a zdroje pro dostatečný sběr dat. Jedinými výjimkami z procesu PMA v rámci třídy III jsou zařízení s podstatným ekvivalentem. Můžete určit, zda zařízení třídy III může být na trh s 510 (k) hledáním FDA Premarket schválení(PMA) databáze a 510 (k) databáze oznámení Premarket.,
jak určit svou třídu
prvním krokem k klasifikaci vašeho zdravotnického prostředku je navigace v klasifikačních předpisech FDA, seznamu 16 kategorií zdravotnických prostředků podle lékařské specializace.
jako příklad vám ukážeme kroky k identifikaci klasifikace poplachu krevního tlaku. Přístroj je zařazen do kategorie 870: kardiovaskulární zařízení.,
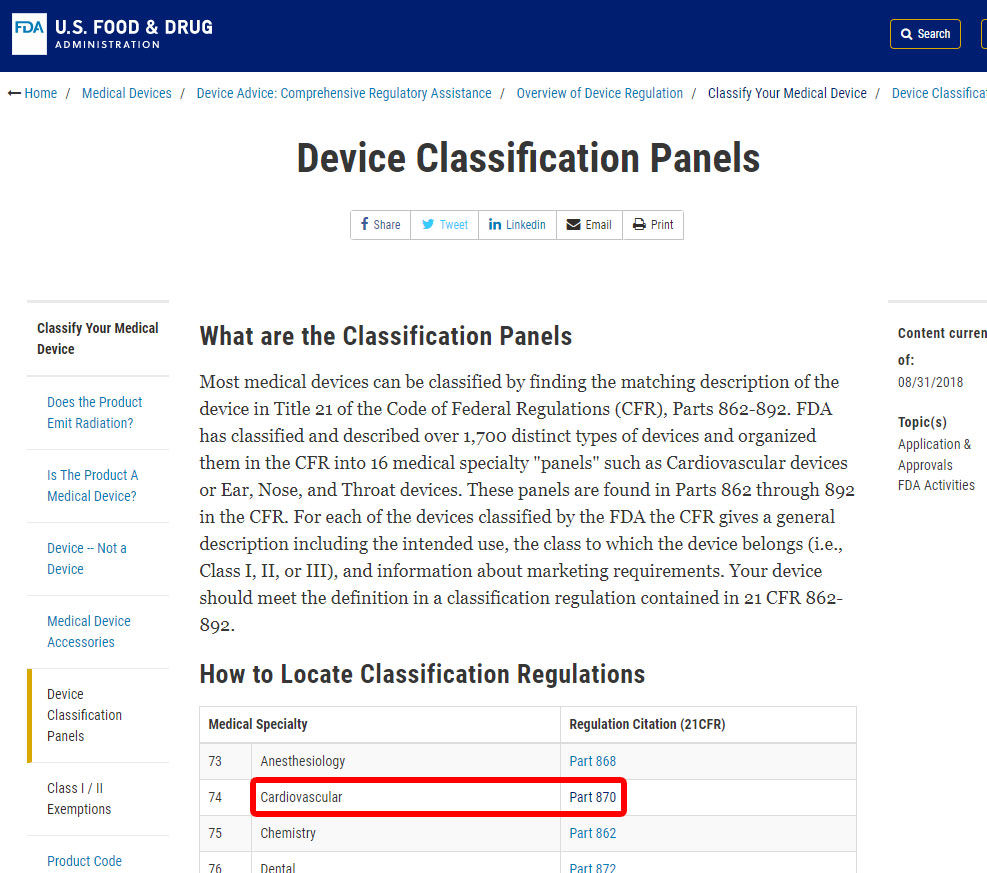
Jakmile jste“ve nachází příslušného lékařského oboru, klepněte na kategorii, a přejděte na seznam zařízení, dokud nenajdete odpovídající a související zařízení kód.
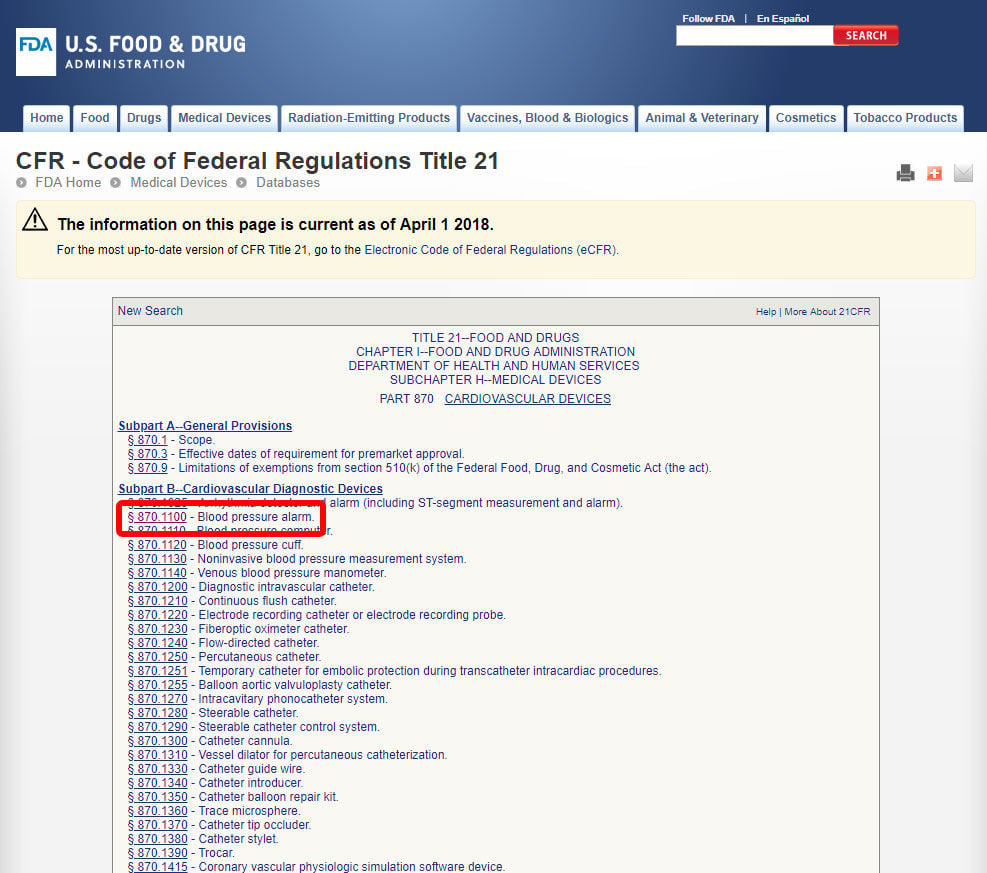
klikněte na kód zařízení a otevřete pokyny. Klasifikace zařízení je uvedena v části b).,
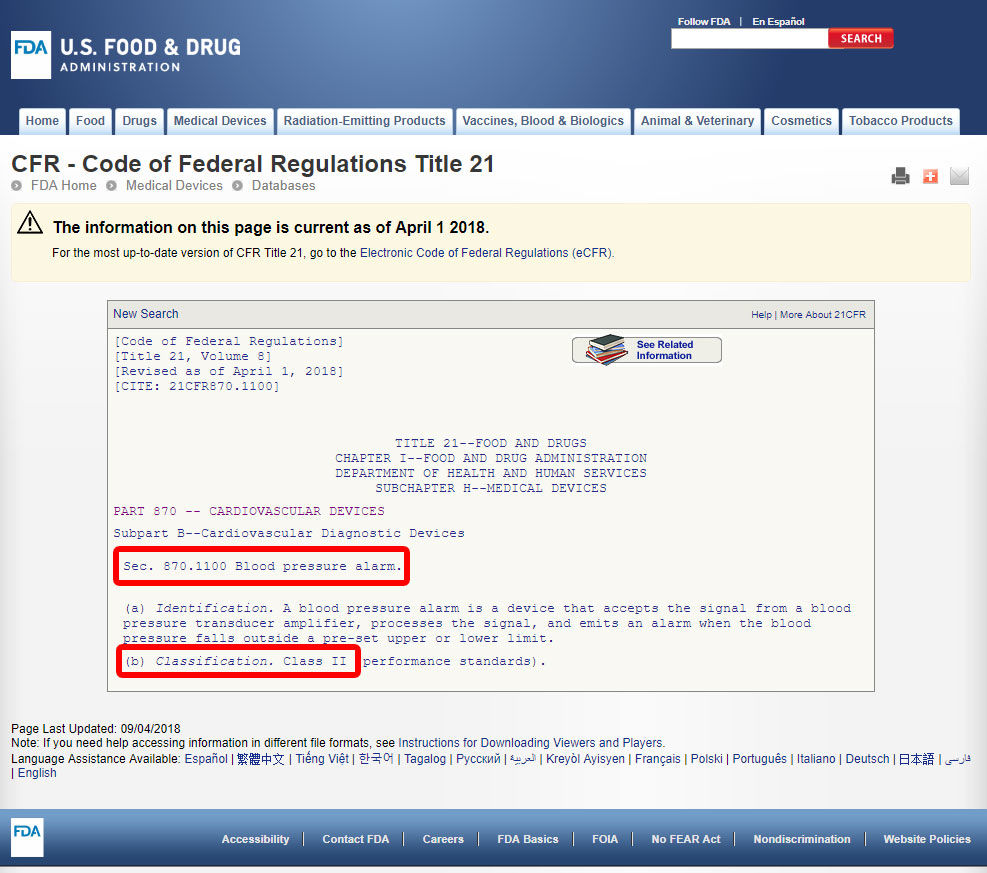
Pokud vaše zařízení nemá uvedené rovnocenné mezi 1700 zařízení klasifikovány podle FDA, je velmi pravděpodobné, inovativní zařízení bez podstatného ekvivalentní a měl by být klasifikován jako Třída III.
Pochopení FDA Lékařské Zařízení Třídy
rozdíly mezi zdravotnických prostředků klasifikovaných jako Třída I, II, nebo III FDA je většinou riziko, množství kontaktu s pacientem a jejich interními systémy, a zda zařízení je rozhodující pro udržení života.,
kromě těchto faktorů považuje FDA za podstatnou rovnocennost při určování toho, jak je zařízení klasifikováno. Pokud je vaše zařízení s nízkým rizikem a minimálně kontaktuje pacienta, pravděpodobně se kvalifikujete pro třídu I a zjednodušený proces schvalování trhu. Zařízení třídy II a III musí prokázat bezpečnost prostřednictvím hmotné rovnocennosti, podání 510(k) nebo procesu schvalování premarketu.
tím, že víte, jak je vaše zařízení klasifikováno, můžete zefektivnit cestu ke schválení trhu pochopením procesů a dokumentů, které pravděpodobně FDA vyžaduje., Pokud vaše organizace podléhá požadavku třídy II nebo třídy III 510 (k) nebo PMA, mohou vám tyto znalosti pomoci předem přidělit příslušné zdroje a naplánovat úspěšné podání.
