Care sunt diferențele în clasele de dispozitive medicale FDA?
FDA din SUA reglementează toate dispozitivele medicale comercializate în SUA, care sunt grupate în trei clase largi. Orice dispozitiv medical aprobat de FDA este clasificat fie ca clasa I, II sau III, în funcție de riscul dispozitivului, invazivitatea și impactul asupra sănătății generale a pacientului. Dar unde sunt liniile trasate între fiecare dintre aceste trei clase și de ce?,liniile directoare de clasificare ale FDA din SUA pot fi foarte confuze pentru producătorii de dispozitive medicale cu expunere limitată la sistem. Există o diferență enormă în calea optimă către piață pentru producători, în funcție de modul în care este grupat dispozitivul. Dispozitivele din clasa I sunt supuse unor cerințe de reglementare mult mai puține decât dispozitivele din clasa II sau III.înțelegând diferențele dintre clasele de dispozitive medicale FDA, puteți înțelege modul în care dispozitivul dvs. va fi grupat., Cu aceste cunoștințe în mână, producătorii de dispozitive medicale din etapele premarket pot pregăti și aloca mai bine resursele necesare aprobării de reglementare.
diferențele dintre clasele de dispozitive medicale FDA
FDA a clasificat peste 1,700 de tipuri distincte de dispozitive medicale. Dispozitivele sunt organizate în codul reglementărilor federale (CFR) în conformitate cu 16 specialități, cum ar fi dispozitivele cardiovasculare sau hematologice., Clasificarea dispozitivului medical în funcție de una dintre cele 16 specialități este primul pas pentru a înțelege dacă fabricați un dispozitiv medical de clasa I, II sau III.după clasificarea unui dispozitiv în funcție de specialitate, FDA instruiește producătorii să procedeze la notificarea premarket cu cunoașterea faptului dacă dispozitivul lor este scutit sau nu. Dispozitivele medicale din clasa I, categoria Cea mai puțin riscantă și invazivă, sunt scutite de procesele de notificare premarket. Dispozitivele specifice din clasa II sunt, de asemenea, scutite de aprobarea premarket.,cu toate acestea, toate dispozitivele reglementate de FDA sunt supuse cerințelor actuale de bună practică de fabricație (cGMP) pentru înregistrare, etichetare și calitate. Dar cum știi dacă dispozitivul este clasa I sau II, și dacă sunteți obligat să se supună notificare premarket? 
Clasa 1
FDA SUA definește Clasa I aparate și dispozitive care „nu sunt destinate utilizării în sprijin sau de susținere a vieții sau de o importanță deosebită în prevenirea afectare a sănătății umane, și ei nu pot prezenta un potențial risc nerezonabile de boală sau rănire.,aceste dispozitive sunt cea mai comună clasă de dispozitive reglementate de FDA, constituind 47% Din dispozitivele aprobate de pe piață.dispozitivele din clasa I au un contact minim cu pacienții și un impact redus asupra sănătății generale a pacientului. În general, dispozitivele din clasa I nu intră în contact cu organele interne ale pacientului, cu sistemul nervos central sau cu sistemul cardiovascular. Aceste dispozitive sunt supuse celor mai puține cerințe de reglementare.,
Exemple de Clasa I Dispozitive:
- Periuta de dinti Electrica
- Apăsător de Limbă
- Masca de Oxigen
- Reutilizabile Bisturiu Chirurgical
- Bandaje
- Paturi de Spital
Aducerea Clasa I a Dispozitivelor Medicale de pe Piață
Dispozitivele din clasa I sunt cel mai rapid și simplu pentru a aduce pe piață deoarece acestea prezintă cea mai mică cantitate de risc pentru pacient și sunt rareori critică pentru susținerea artificială a vieții. Majoritatea dispozitivelor din clasa I sunt scutite de cerințele FDA pentru Notificarea Premarket (510k) și aprobarea Premarket (PMA).,dispozitivele din clasa I nu sunt scutite de controalele generale FDA, o serie de comenzi care se aplică dispozitivelor medicale din clasa I, II și III. Prevederile acestui act se referă la falsificare, etichetare greșită, înregistrarea dispozitivelor, înregistrări și bune practici de fabricație. Producătorii de dispozitive medicale care se încadrează în clasa A sunt în continuare obligați să implementeze un sistem de management al calității și să respecte standardele pentru a asigura un produs de calitate.,
lectură înrudită: diferența dintre Notificarea Premarket 510(k) și aprobarea Premarket
clasa 2
dispozitivele medicale din clasa II sunt mai complicate decât dispozitivele din clasa I și prezintă o categorie de risc mai mare, deoarece sunt mai susceptibile de a intra în contact susținut cu un pacient. Aceasta poate include dispozitive care vin în contact cu sistemul cardiovascular al pacientului sau cu organele interne și instrumente de diagnosticare.,FDA definește dispozitivele din clasa II ca ” dispozitive pentru care controalele generale sunt insuficiente pentru a oferi o asigurare rezonabilă a siguranței și eficacității dispozitivului.,est Kituri
Aducerea Clasa a II-a a Dispozitivelor Medicale de pe Piață
Controale varia în funcție de dispozitiv, dar, potrivit FDA, pot include:
- Dispozitiv de performanță
- de supraveghere ulterioară introducerii pe piață
- registre de pacienți
- Special cerințele de etichetare
- Premarket cerințele de date
- Directoare
majoritatea de Clasa a II-a de dispozitive sunt aprobate de FDA pentru piață prin Pre-Notificare, sau 510(k) proces.,dispozitivele din clasa II sunt supuse acelorași controale generale menționate mai sus, dar FDA le definește ca fiind „dispozitive pentru care controalele generale sunt insuficiente pentru a oferi o asigurare rezonabilă a siguranței și eficacității dispozitivului.”Din acest motiv, dispozitivele din clasa II sunt, de asemenea, supuse unor controale speciale. Aceste reglementări depind de dispozitiv și pot include cerințe speciale de etichetare, registre ale pacienților și standarde de performanță.
majoritatea dispozitivelor din clasa II vin pe piață folosind procesul de notificare Premarket (510K)., 510 (k) este o aplicație complexă pentru FDA, care demonstrează că un dispozitiv este sigur și eficient, demonstrând că dispozitivul este echivalent cu un alt dispozitiv care este pe piață.acest proces implică afișarea ” echivalenței substanțiale „cu un alt dispozitiv care este cunoscut în limbajul FDA ca” predicatul.”Acest lucru nu înseamnă că dispozitivele trebuie să fie identice, dar necesită asemănări semnificative în utilizare, design, materiale, etichetare, standarde și alte caracteristici.,FDA a lansat o listă de scutiri la începutul anului 2018, care scutește peste dispozitivele medicale generice 800 din clasa I și II de la procesul 510(k). Dacă aveți un dispozitiv medical generic de clasa II, puteți descoperi dacă acesta este scutit de o depunere de 510(k) prin căutarea în baza de date de clasificare a produselor FDA.
lectură înrudită: 5 motive revizuirea FDA 510 (k) este o mișcare excelentă.FDA definește dispozitivele din clasa III ca produse care ” de obicei susțin sau susțin viața, sunt implantate sau prezintă un risc potențial nerezonabil de boală sau rănire.,doar 10% din dispozitivele reglementate de FDA din SUA se încadrează în clasa III. această clasificare este în general extinsă la implanturi permanente, dispozitive medicale inteligente și sisteme de susținere a vieții.în timp ce clasa III este în general rezervată celor mai inovatoare și de ultimă oră dispozitive medicale, există și alte dispozitive care pot intra în clasa III din diferite motive., Unele dispozitive care sunt clasificate inițial ca clasa II pot fi lovite până la clasa III dacă producătorul nu poate demonstra echivalența substanțială cu un predicat (produs existent) în timpul procesului de depunere PMA (510k).,
Exemple de dispozitive medicale din clasa III:
- implanturi mamare
- stimulatoare cardiace
- defibrilatoare
- ventilatoare de înaltă frecvență
- implanturi cohleare
- monitoare de prelevare a sângelui Fetal
- proteze implantate
aducerea pe piață a Dispozitivelor Medicale din clasa III
dispozitivele din clasa procesul de aprobare premarket (PMA)., FDA scrie: „datorită nivelului de risc asociat dispozitivelor din clasa III, FDA a stabilit că controalele generale și speciale sunt insuficiente pentru a asigura siguranța și eficacitatea dispozitivelor din clasa III.”
PMA este cel mai intens tip de aplicație de marketing dispozitiv cerut de FDA. Unele dispozitive FDA clasa III sunt scutite și se pot califica pentru o depunere de 510(k), dar majoritatea sunt de așteptat să obțină aprobarea Premarket.,procesul PMA necesită un studiu riguros al unui dispozitiv medical pentru a dovedi siguranța și eficacitatea prin dezvoltarea unui profil beneficiu/risc bazat pe date. Procesul PMA implică, în general, studii clinice și timp și resurse semnificative pentru colectarea suficientă a datelor. Singurele excepții de la procesul PMA din clasa III sunt dispozitivele cu un echivalent substanțial. Puteți determina dacă un dispozitiv de clasa III poate fi comercializat cu un 510(k) căutând baza de date FDA Premarket Approval (PMA) și baza de date de notificare Premarket 510(k).,primul pas spre clasificarea dispozitivului Medical este de a naviga reglementările de clasificare FDA, lista de 16 categorii pentru dispozitive medicale în funcție de specializare medicală.de exemplu, vă vom arăta pașii pentru identificarea clasificării unei alarme a tensiunii arteriale. Dispozitivul este clasificat în categoria 870: dispozitive cardiovasculare.,după ce ați localizat specialitatea medicală relevantă, faceți clic pe categorie și navigați în lista dispozitivelor până când găsiți un echivalent și Codul dispozitivului asociat.
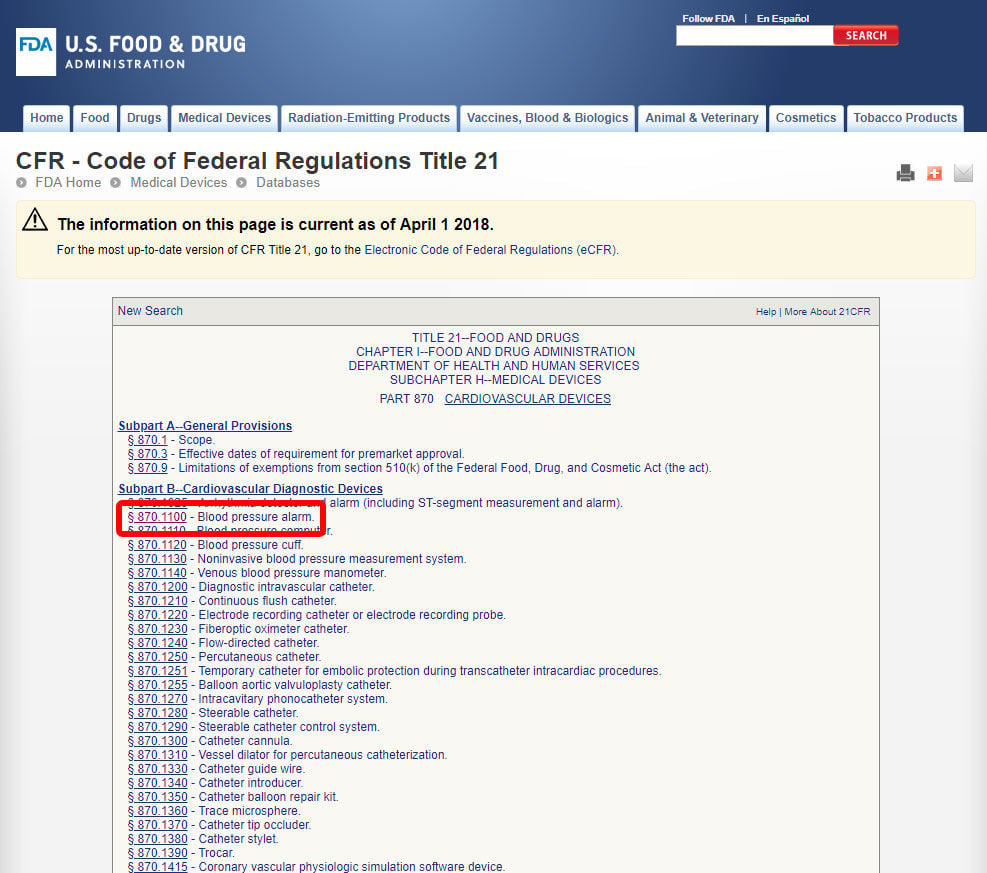
Faceți clic pe codul dispozitivului și deschideți liniile directoare. Clasificarea dispozitivelor este prezentată în secțiunea (b).,
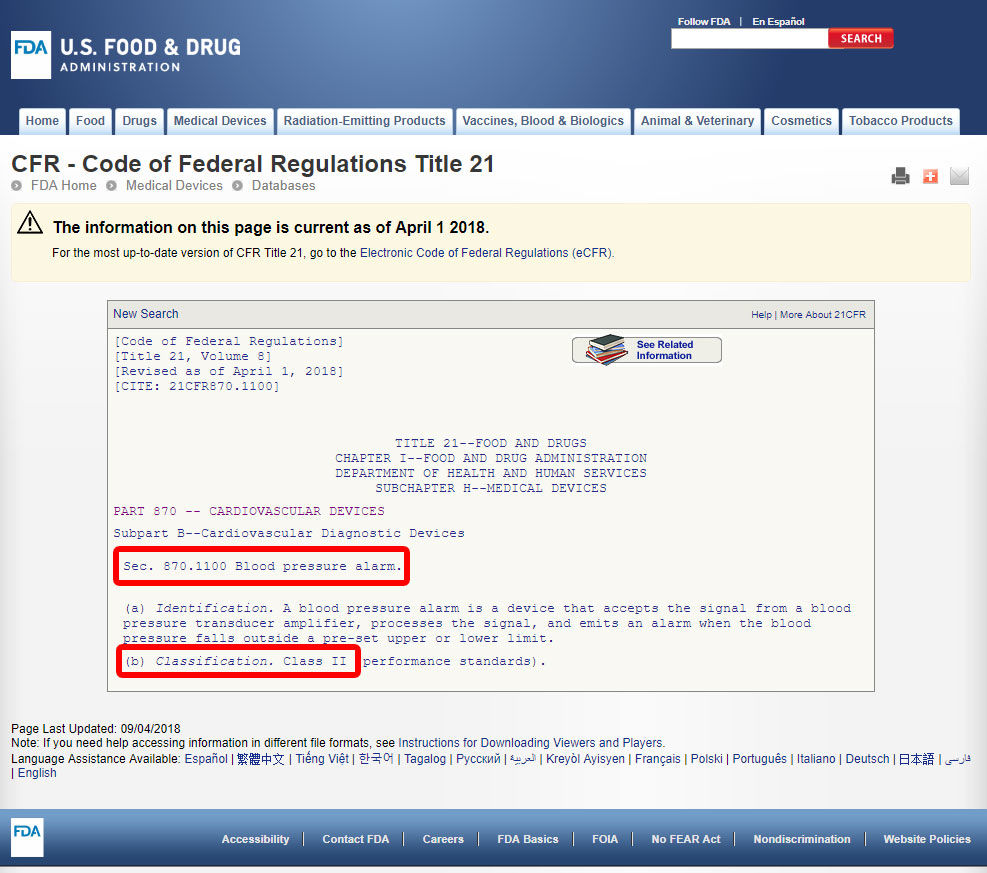
Dacă dispozitivul nu are un enumerate echivalente între 1.700 de dispozitive clasificate în funcție de FDA, acesta este cel mai probabil un dispozitiv inovator fără o substanțială echivalent și vor fi încadrate în Clasa a III-a.
Înțelegere FDA Dispozitiv Medical Clase
diferențele între dispozitivele medicale din Clasa I, II, sau III de FDA este cea mai mare de risc, suma de contact cu un pacient și sistemele lor interne, și dacă un dispozitiv este esențială pentru a susține viața.,în plus față de acești factori, FDA consideră echivalență substanțială atunci când determină modul în care este clasificat un dispozitiv. Dacă dispozitivul dvs. prezintă un risc scăzut și contactează minim pacientul, este posibil să vă calificați pentru clasa I și un proces simplificat de aprobare pe piață. Dispozitivele din clasele II și III trebuie să demonstreze siguranța prin echivalență de fond, printr-o depunere de 510(k) sau prin procesul de aprobare premarket.cunoscând modul în care este clasificat dispozitivul dvs., vă puteți eficientiza calea către aprobarea pieței înțelegând procesele și documentele care ar putea fi solicitate de FDA., Dacă organizația dvs. este supusă unei cerințe Clasa II sau clasa III 510(k) sau PMA, aceste cunoștințe vă pot ajuta să alocați resursele corespunzătoare în avans și să planificați o depunere reușită.
