Hvad er forskellene i FDA Medical Device klasser?
den amerikanske FDA regulerer alt medicinsk udstyr, der markedsføres i USA, og som er grupperet i tre brede klasser. Enhver medicinsk udstyr, der er godkendt af FDA er klassificeret som enten klasse I, II, eller III afhængigt af enheden”s risiko, invasivitet, og indvirkning på patientens generelle sundhed. Men hvor er linjerne trukket mellem hver af disse tre klasser, og hvorfor?,de amerikanske FDA ” s-klassificeringsretningslinjer kan være meget forvirrende for producenter af medicinsk udstyr med begrænset eksponering for systemet. Der er en enorm forskel i den optimale vej til markedet for producenter afhængigt af, hvordan din enhed er grupperet. Klasse i-enheder er underlagt langt færre lovkrav end klasse II-eller III-enheder.
Ved at forstå forskellene i FDA ‘ s medicinske udstyrsklasser kan du forstå, hvordan din enhed vil blive grupperet., Med denne viden i hånden kan producenter af medicinsk udstyr i premarket-stadierne bedre forberede og allokere de ressourcer, der er nødvendige til lovgivningsmæssig godkendelse.
forskelle mellem FDA Medical Device Classes
FDA har klassificeret over 1.700 forskellige typer medicinsk udstyr. Enhederne er organiseret i Code of Federal Regulations (CFR) i henhold til 16 specialiteter, såsom kardiovaskulære eller hæmatologiske enheder., Klassificering af dit medicinske udstyr i henhold til en af de 16 specialiteter er det første skridt til at forstå, om du fremstiller et medicinsk udstyr i klasse I, II eller III.
efter klassificering af en enhed i henhold til specialitet instruerer FDA producenterne om at gå videre til forhåndsmeddelelse med viden om, hvorvidt deres enhed er fritaget eller ej. Medicinsk udstyr i klasse I, den mindst risikable og invasive kategori, er fritaget for underretningsprocesser før markedsføring. Specifikke klasse II-enheder er også undtaget fra forhåndsgodkendelse.,
alle enheder, der reguleres af FDA, er dog underlagt de nuværende cGMP-krav (Good Manufacturing Practice) til registrering, mærkning og kvalitet. Men hvordan kan du vide, om din enhed er klasse i eller II, og om du er forpligtet til at gennemgå forudgående meddelelse? 
Klasse 1
Det AMERIKANSKE FDA definerer Klasse i udstyr som enheder, der “ikke beregnet til brug i støtte eller opretholdelse af livet eller af væsentlig betydning for at forebygge forringelse for menneskers sundhed, og de kan ikke udgøre en potentiel urimelig risiko for sygdom eller skade.,”
disse enheder er den mest almindelige klasse af enheder reguleret af FDA, der udgør 47 procent af godkendte enheder på markedet.
klasse i-enheder har minimal kontakt med patienter og lav indvirkning på en patient”s generelle sundhed. Generelt behøver klasse I-enheder ikke komme i kontakt med en patient”s indre organer, centralnervesystemet, eller kardiovaskulære system. Disse enheder er underlagt de færreste lovkrav.,
Eksempler på Udstyr i Klasse i:
- Elektrisk Tandbørste
- tungespatler
- Oxygen Maske
- Genanvendeligt Kirurgisk Skalpel
- Bandager
- hospitalssenge
Bringe Klasse i Medicinsk Udstyr på Markedet
Udstyr i klasse i er den hurtigste og nemmeste at bringe til markedet, da de udgør det laveste beløb af risiko for patienten og er sjældent kritisk til livsforlængende behandling. Størstedelen af klasse I-enheder er undtaget fra FDA-krav til forhåndsmeddelelse (510k) og forhåndsgodkendelse (PMA).,
klasse i-enheder er ikke undtaget fra FDA ‘ s generelle kontroller, en række kommandoer, der gælder for klasse I, II og III medicinsk udstyr. Bestemmelserne i denne lov vedrører forfalskning, misbranding, enhedsregistrering, poster, og god fremstillingspraksis. Producenter af medicinsk udstyr, der falder i klasse A, er stadig forpligtet til at implementere et kvalitetsstyringssystem og følge standarder for at sikre et kvalitetsprodukt.,
relateret læsning: forskellen mellem meddelelse før markedsføring 510(k) og godkendelse før markedsføring
klasse 2
klasse II medicinsk udstyr er mere kompliceret end klasse i-udstyr og udgør en højere kategori af risiko, fordi de er mere tilbøjelige til at komme i vedvarende kontakt med en patient. Dette kan omfatte enheder, som kommer i kontakt med en patient”s kardiovaskulære system eller indre organer, og diagnostiske værktøjer.,
FDA definerer klasse II-enheder som “enheder, for hvilke generelle kontroller ikke er tilstrækkelige til at give rimelig sikkerhed for enhedens sikkerhed og effektivitet.,est Kits
Bringe Klasse II Medicinsk Udstyr på Markedet
Kontrol variere afhængigt af enheden, men i henhold til FDA, kan nævnes:
- Enhedens ydeevne
- Postmarket overvågning
- patientregistre
- Særlige mærkningskrav
- før markedsføring krav til data
- Retningslinjer
størstedelen af Klasse II-udstyr, er FDA godkendt til markedet gennem før markedsføring Meddelelse, eller 510(k) proces.,klasse II-enheder er underlagt de samme generelle kontroller, der er nævnt ovenfor, men FDA definerer dem som værende “enheder, for hvilke generelle kontroller ikke er tilstrækkelige til at give rimelig sikkerhed for enhedens sikkerhed og effektivitet.”Af den grund er klasse II-enheder også underlagt særlige kontroller. Disse regler afhænger af enheden og kan omfatte særlige mærkningskrav, patientregistre og præstationsstandarder.
de fleste klasse II-enheder kommer på markedet ved hjælp af premarket Notification (510k) processen., 510 (k) er en kompleks applikation til FDA, som viser, at en enhed er sikker og effektiv ved at demonstrere, at enheden svarer til en anden enhed, der er på markedet.
denne proces indebærer at vise “væsentlig ækvivalens” til en anden enhed, der er kendt i FDA parlance som “prædikatet.”Dette betyder ikke, at enhederne skal være identiske, men de kræver betydelige ligheder i brug, design, materialer, mærkning, standarder og andre egenskaber.,
FDA frigav en undtagelsesliste i begyndelsen af 2018, der fritager over 800 generiske klasse I og II medicinsk udstyr fra 510(k) – processen. Hvis du har et generisk klasse II-medicinsk udstyr, kan du finde ud af, om det er fritaget for en 510(k) arkivering ved at søge i FDA-Produktklassifikationsdatabasen.
relateret læsning: 5 grunde overhaling FDA 510(k) er et godt træk.
klasse 3
FDA definerer klasse III-enheder som produkter, der “normalt opretholder eller understøtter liv, implanteres eller udgør en potentiel urimelig risiko for sygdom eller skade.,”
kun 10 procent af de enheder, der reguleres af den amerikanske FDA, falder i klasse III. denne klassificering udvides generelt til permanente implantater, smart medicinsk udstyr og livsstøttesystemer.mens klasse III generelt er forbeholdt de mest innovative og banebrydende medicinsk udstyr, er der andre enheder, der kan falde i klasse III af forskellige årsager., Nogle enheder, der oprindeligt kategoriseres som klasse II, kan blive stødt op til klasse III, hvis producenten ikke er i stand til at demonstrere væsentlig ækvivalens med et predikat (eksisterende produkt) under PMA (510k) arkiveringsprocessen.,
Eksempler på Medicinsk Udstyr under Klasse III:
- Brystimplantater
- Pacemakere
- Defibrillatorer
- Høj-frekvens ventilatorer
- Cochlear implantater
- Føtal blodprøvetagning skærme
- Indopereret proteser
Bringe Klasse III Medicinsk Udstyr på Markedet
Class III udstyr er underlagt FDA Generel Kontrol og FDA Godkendelse før markedsføring (PMA) proces., FDA skriver: “på grund af risikoniveauet forbundet med klasse III-enheder har FDA bestemt, at generelle og specielle kontroller alene ikke er tilstrækkelige til at sikre sikkerheden og effektiviteten af klasse III-enheder.”
PMA er den mest intensive type enhedsmarkedsføringsapplikation, der kræves af FDA. Nogle FDA klasse III-enheder er undtaget og kan kvalificere sig til en 510 (k) arkivering, men flertallet forventes at få godkendelse før Market.,
PMA-processen kræver en grundig undersøgelse af et medicinsk udstyr for at bevise sikkerhed og effektivitet gennem udvikling af en datadrevet fordel / risikoprofil. PMA-processen involverer generelt kliniske forsøg og betydelig tid og ressourcer til tilstrækkelig dataindsamling. De eneste undtagelser fra PMA-processen inden for klasse III er enheder med en betydelig ækvivalent. Du kan afgøre, om en Klasse III-enhed, kan markedsføres med en 510(k) ved at søge FDA Godkendelse før markedsføring (PMA) database og 510(k) før markedsføring Anmeldelse database.,
Sådan bestemmer du din klasse
det første skridt mod klassificering af dit medicinske udstyr er at navigere i FDA-Klassificeringsbestemmelserne, listen over 16 kategorier for medicinsk udstyr i henhold til medicinsk specialisering.
som et eksempel viser vi”trinnene til at identificere klassificeringen af en blodtryksalarm. Enheden er klassificeret under kategori 870: kardiovaskulære enheder.,
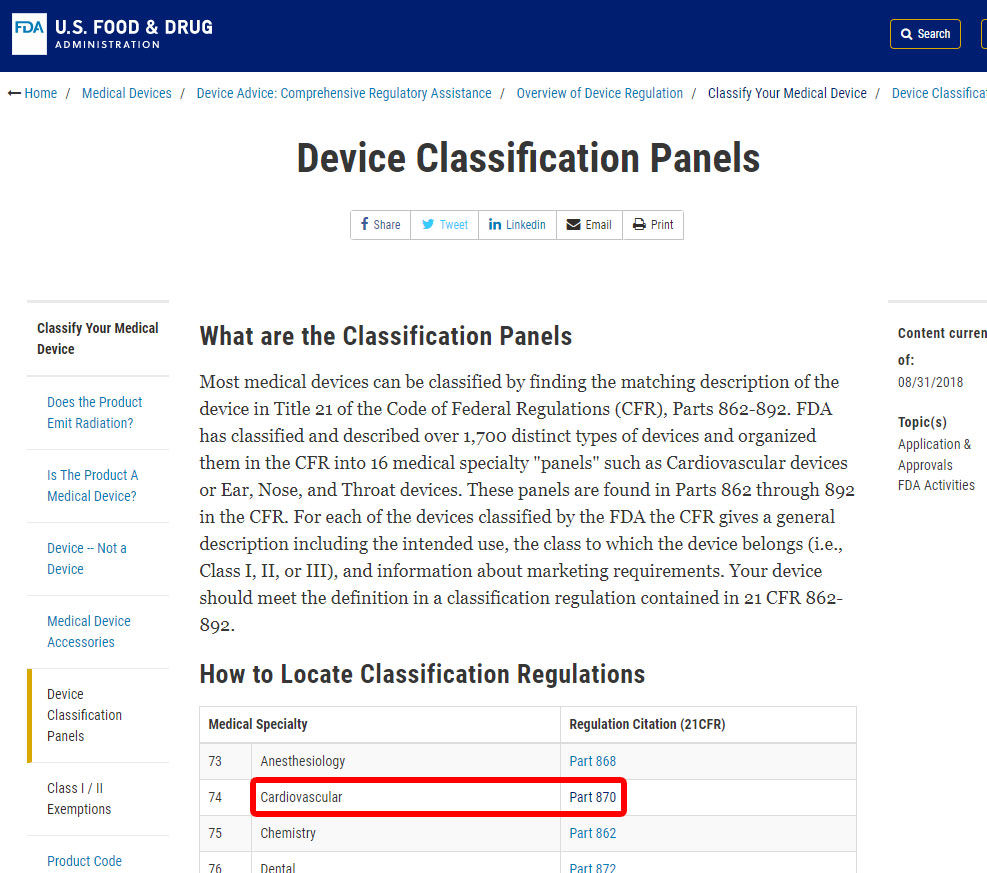
Når du har fundet den relevante medicinske specialitet, skal du klikke på kategorien og navigere i listen over enheder, indtil du finder en tilsvarende og den tilhørende enhedskode.
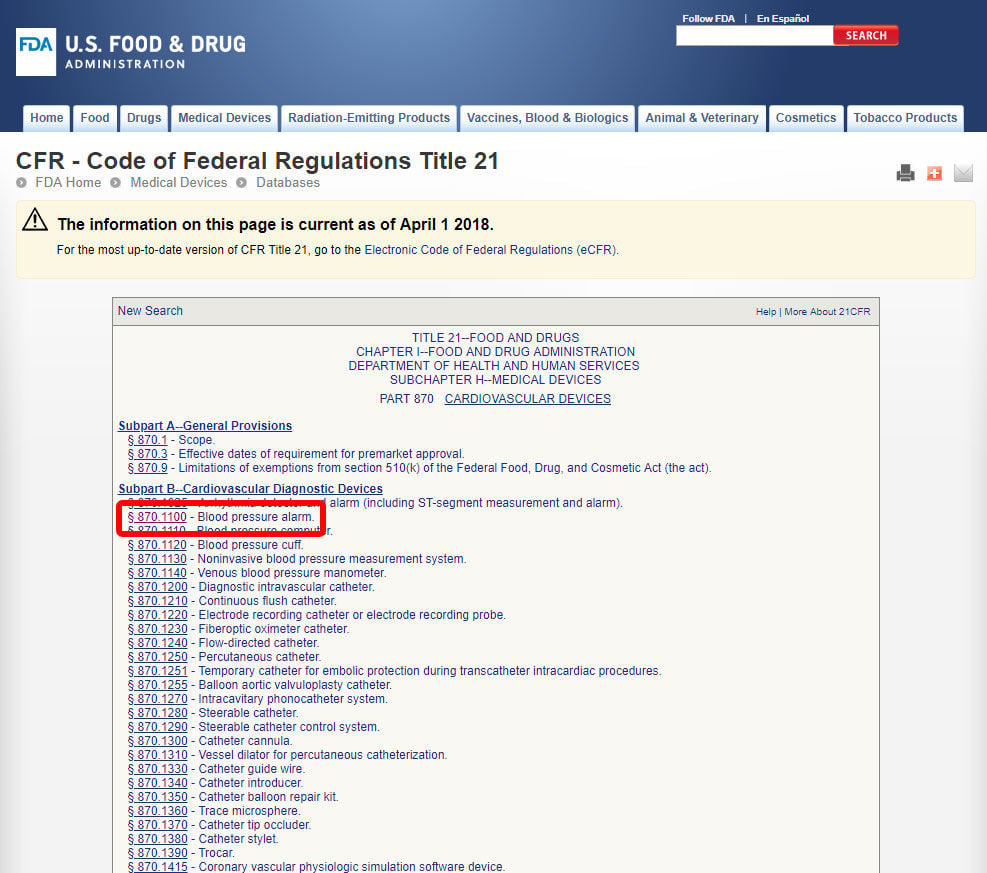
Klik på enhedskoden og åbn retningslinjerne. Enhedsklassifikationen er anført under afsnit b).,
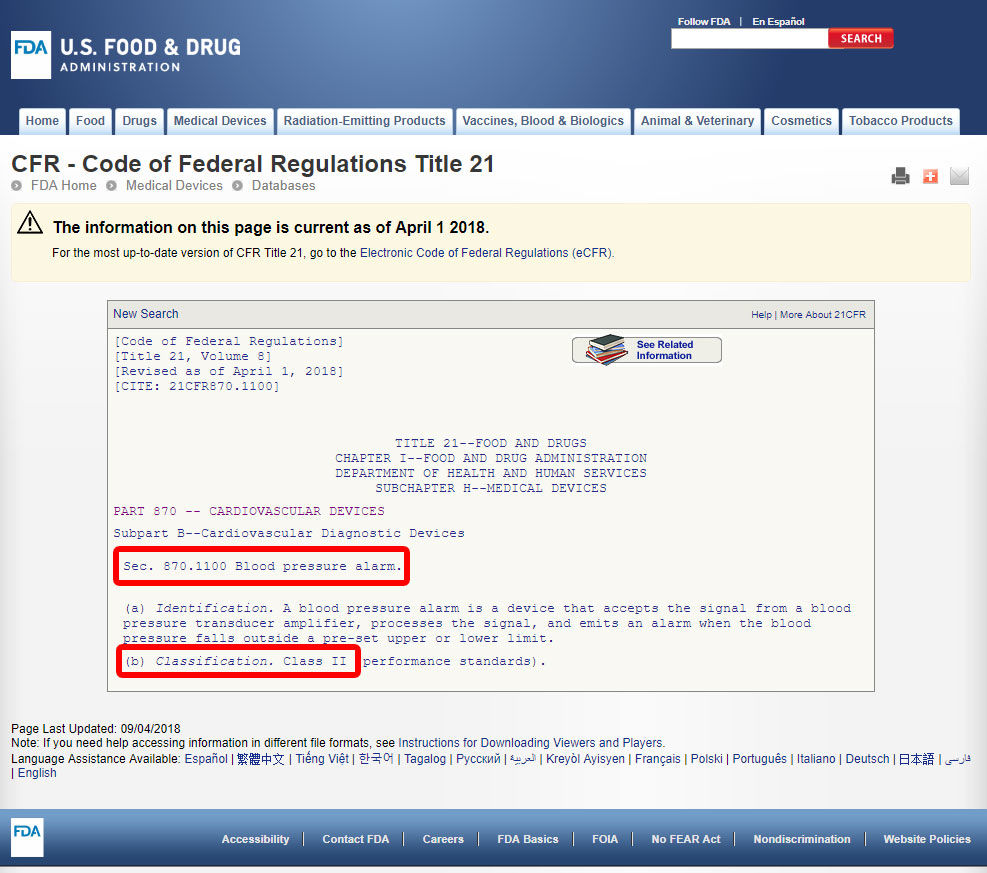
Hvis din enhed ikke har en, der er anført tilsvarende blandt de 1,700 udstyr, der er klassificeret af FDA, er det mest sandsynligt en nyskabende enhed uden en betydelig tilsvarende og vil blive klassificeret som Klasse III.
Forståelse FDA Medicinsk udstyr Klasser
forskellene mellem medicinsk udstyr, der er klassificeret som Klasse i, II, eller III af FDA er for det meste risiko, mængden af kontakt med en patient, og deres interne systemer, og uanset om en enhed er afgørende for opretholdelse af livet.,
ud over disse faktorer overvejer FDA væsentlig ækvivalens, når den bestemmer, hvordan en enhed klassificeres. Hvis din enhed er lav risiko og minimalt kontakter patienten, vil du sandsynligvis kvalificere dig til klasse I og en strømlinet markedsgodkendelsesproces. Udstyr i klasse II og III skal demonstrere sikkerhed via materiel ækvivalens, en 510 (k) arkivering eller godkendelsesprocessen før markedsføring.
Ved at vide, hvordan din enhed er klassificeret, kan du strømline din vej til markedsgodkendelse ved at forstå de processer og dokumenter, som FDA sandsynligvis vil kræve., Hvis din organisation er underlagt et krav i klasse II eller klasse III 510(k) eller PMA, kan denne viden hjælpe dig med at allokere de relevante ressourcer på forhånd og planlægge en vellykket arkivering.
