Holoprosenzephalie und Strabismus
Pavlina S. Kemp, MD, Grant Casey, Susannah Q. Longmuir, MD
12. Juni 2012
Hauptbeschwerde: Augenkreuzung
Vorgeschichte der vorliegenden Erkrankung
Die Patientin ist eine 15 Monate alte Frau bei der Präsentation in der Augenklinik, mit der Vorgeschichte eines schweren Hydrozephalus bei der Geburt. Sie wurde auch mit alobar Holoprosencephalie bei der Geburt mit Anfällen diagnostiziert. Sie wurde ursprünglich für Eye Crossing bezeichnet. Wir präsentieren ihren bemerkenswerten klinischen Verlauf.,
Vergangene Krankengeschichte:
- Hydrocephalus s/p Ventrikuloperitoneal Shuntting
- Holoprosencephalie (Alobar-Typ)
- Anfallsstörung
Vergangene chirurgische Vorgeschichte:
- Ventrikuloperitoneal Shunt Placement, 2004
- Ventrikuloperitoneal shunt revision, 1/2005
- li> Ventrikuloperitoneal shunt revision, 6/2005
Familienanamnese: Keine bekannte Familienanamnese von Holoprosenzephalie, Amblyopie oder Strabismus.
Sozialgeschichte: Patient lebt zu Hause mit Eltern und zwei Schwestern.,
Medikamente: Keine
Untersuchung und klinischer Verlauf:
Alter: 15 Monate
Sehschärfe: Zentral, instabil und aufrechterhalten OD und zentral, instabil und aufrechterhalten OS
Teller acuity testing:
- Ohne Korrektur OU: 20/800
Pupillen: Gleich rund und lebhaft reaktiv, kein relativer afferenter Pupillendefekt.,
Stereo Vision: Unfähig zu testen
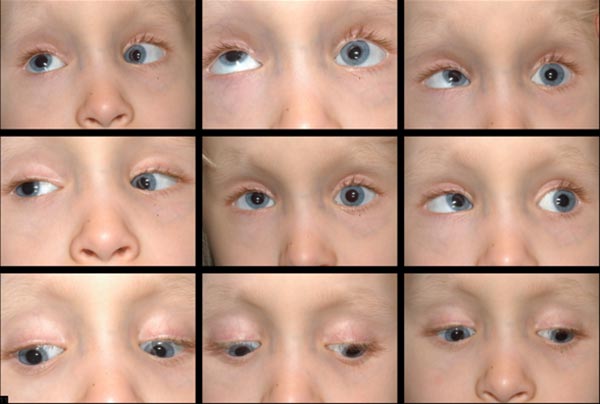
Motilität und Strabismus:
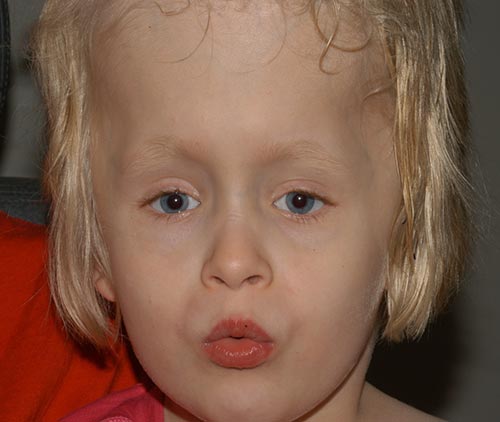
- Große variable Esotropie
- Bilaterale Elevations-und Abduktionsdefizite
- Intermittierender horizontaler Nystagmus
Cycloplegische Refraktion:
- OD: +4.00
- OS: +6.00
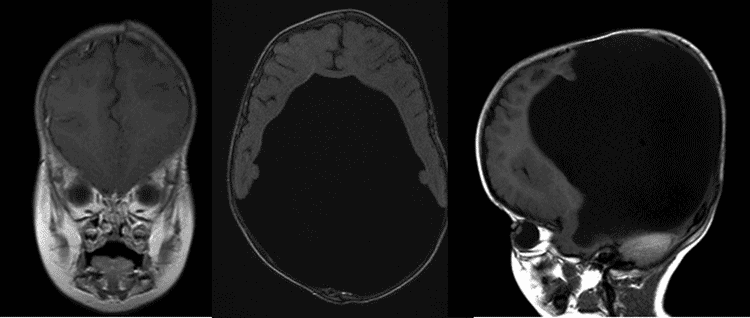
Externe Untersuchung: Bemerkenswert für großen Kopfumfang
Spaltlampenuntersuchung: Normale Untersuchung des vorderen Segments OU ohne Anzeichen von Katarakten oder anderen Medientrübungen.
Normal erscheinende Sehnerven und normale erweiterte Fundusuntersuchung. Keine Anzeichen von Sehnervenhypoplasie in beiden Augen.,
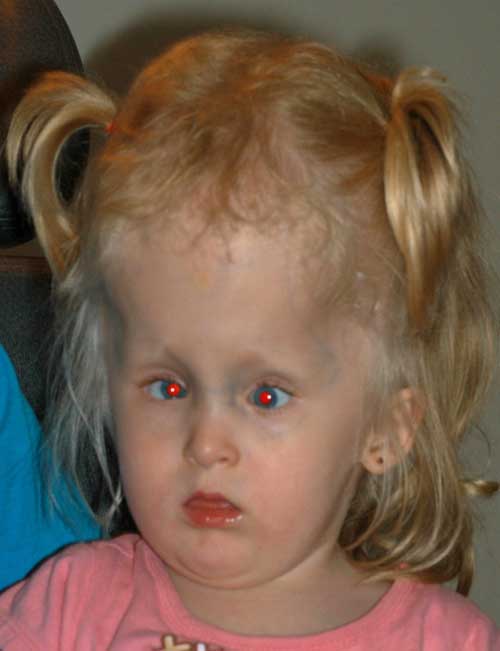
Zu diesem Zeitpunkt wurde nach Gesprächen mit ihrer Familie eine Operation wegen Strabismus verschoben und eine Korrektur ihrer Hyperopie wurde versucht. Gläser wurden verschrieben. Sie konnte keine Brille bequem tragen und Kontaktlinsen wurden ausprobiert. Patching wurde durchgeführt, um ihre Amblyopie zu behandeln. Die Patientin wurde verfolgt und im Alter von 3 Jahren wollten ihre Eltern mit einer Strabismusoperation fortfahren, um ihre Augen zu „kreuzen“.,
Alter: 3
Sehschärfe: Zentral, CUSM OD und CUSM OS
Motilität und Strabismus:
- Große Winkelesotropie von 55-60 Prismendioptrien (Abbildung 2)
- Bilaterale Abduktionsdefizite und ein linker monokularer Elevationsmangel
Zum Zeitpunkt der Strabismusoperation zeigten intraoperative Zwangsduktionen Einschränkungen sowohl der medialen Rektusmuskulatur als auch des Patienten wurde festgestellt, dass der mediale Rektusmuskel anomal eingeführt wurde. Die mediale Rektusmuskelinsertion vor der Disinsertion wurde 7 mm vom Limbus entfernt gefunden (posteriorer als die erwarteten 5,5 mm vom Limbus entfernt). Da die Anatomie nicht normal war, wurde ein konservativer Ansatz durchgeführt und sie unterzog sich bilateralen medialen Rektusrezessionen von 5, 5 mm, wobei der mediale Rektus bei 12 zurückblieb.,5 mm vom limbus.
Postoperativ hatte sie eine kleine restvariable Esotropie mit geringer vertikaler Abweichung und einem deutlicheren Elevationsmangel des linken Auges.
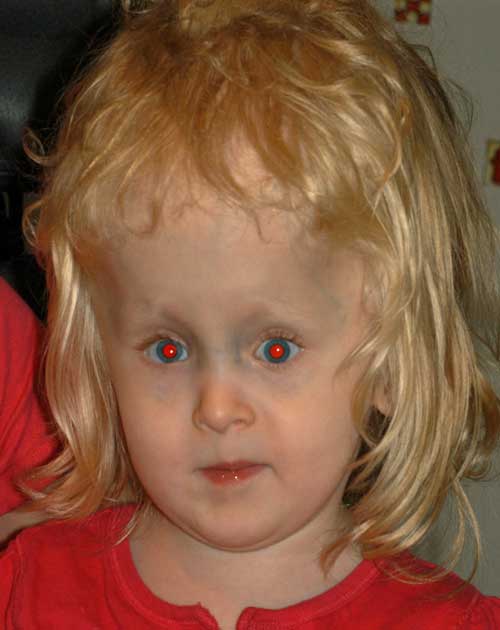
Alter 6:
Ihre Eltern waren mit der Ausrichtung zufrieden; Mit der Zeit entwickelte sie jedoch eine merkliche, rechte Hypertropie. Im Alter von 6 Jahren beschloss ihre Familie, mit einer zweiten Strabismus-Operation fortzufahren, um die vertikale Fehlausrichtung anzugehen.,
Eine Rezession des linken unteren Rektus im Vergleich zu einer Rezession des rechten oberen Rektus war geplant, wobei entschieden wurde, welches Verfahren auf der Grundlage intraoperativer Zwangsduktionen durchgeführt werden würde. Intraoperative Zwangsduktionen zeigten einen engen linken unteren Rektus, was mit dem zuvor in Betracht gezogenen monokularen Elevationsmangel übereinstimmt. Es wurde eine Rezession des linken unteren Rektus von 6 mm durchgeführt, wobei der Muskel von 8 mm posterior zum Limbus, wo er gefunden wurde, auf 14 mm posterior zum Limbus bewegt wurde.,
Einige Monate nach ihrer zweiten Operation bemerkte ihre Mutter, dass ihr rechtes Auge in Zeiten der Unaufmerksamkeit nach oben driftete. Sie wurde in der Klinik gesehen und bemerkte, dass sie eine offensichtliche dissoziierte vertikale Abweichung auf der rechten Seite hatte., Es wurde beschlossen, erneut mit einer Strabismusoperation und einer 8 mm rechten oberen Rektusrezession nach der Hangback-Methode fortzufahren und den Muskel von 8 mm posterior zum Limbus, wo er gefunden wurde, auf 16 mm posterior zum Limbus zu bewegen.
Die Patientin hat sich postoperativ gut entwickelt (Abbildung 6) und ihre visuellen Fähigkeiten schrittweise weiterentwickelt. Sie hat es insgesamt bemerkenswert gut gemacht und geht und liest jetzt.
Diagnose:
Holoprosenzephalie mit infantiler Esotropie und komplexem Strabismus mit Variabilität der medialen Rektusmuskeleinfügungen.
Diskussion:
Die Holoprosenzephalie ist eine Art von kopffüßer Störung, die durch das Versagen des Prosencephalons (des embryonalen Vorderhirns) gekennzeichnet ist und zu einer einklappigen Gehirnstruktur und schweren Schädel-und Gesichtsdefekten führt., Es gibt drei Klassifikationen der Holoprosenzephalie: Alobar holoprosencephaly, semi-lobar holoprosencephaly und lobar holoprosencephaly.
Die alobare Holoprosenzephalie macht zwei Drittel der betroffenen Patienten aus und ist die schwerwiegendste Form, die durch das Versagen des Gehirns gekennzeichnet ist, sich in zwei Hälften zu trennen. Dies führt zu einem einzigen primitiven Ventrikel, fehlenden Geruchszwiebeln und Sehbahnen und schweren Entwicklungsstörungen. Es ist normalerweise mit schweren Gesichtsanomalien verbunden, einschließlich eng beieinanderliegender Augen, kleiner Kopfgröße, Lippen-und Gaumenspalte., Semi-lobare Holoprosenzephalie, die ein Viertel der Fälle von Holoprosenzephalie ausmacht, ist eine intermediäre Form der Erkrankung und zeichnet sich durch teilweise getrennte Gehirnhälften und einen einzelnen Ventrikel aus. Lobare Holoprosenzephalie ist die am wenigsten schwere Form, in der das Gehirn des Patienten fast normal sein kann; Es gibt eine deutliche Fissur zwischen entwickelten Zentrallappen, und eine gewisse Fusion der Gehirnstrukturen ist vorhanden (Nanni, 2000). In den meisten Fällen von Holoprosenzephalie sind Fehlbildungen des Gehirns nicht mit dem Leben vereinbar., In weniger schweren Fällen werden Babys mit normaler oder nahezu normaler Gehirnentwicklung und unterschiedlichem Grad an Gesichtsdeformität geboren.
Midline-kraniofaziale Defekte sind das Markenzeichen der Holoprosenzephalie und können Mikrozephalie, Hypotelorismus (abnormal eng beieinander liegende Augen), Nasenanomalien wie Nasenabflachung oder eine einzelne Naris sowie Oberlippe-und Gaumendefekte wie Gaumenspalte oder einen einzelnen vorderen Schneidezahn umfassen. Zyklopie kann in den schwersten Formen vorhanden sein, in denen ein nasenartiger Rüssel über einem einzelnen Auge in der Mitte des Gesichts vorhanden ist (Nanni, 2000)., Es wird angenommen, dass der Grad der Gesichtsdeformität die Schwere intrakranieller Defekte anzeigt. Assoziierte Komorbiditäten umfassen Funktionsstörungen der Hypophyse und des Hypothalamus, was zu Dysregulation der Körpertemperatur, Anfällen und geistiger Behinderung unterschiedlicher Schwere führt (Dubourg, 2007). Hypotonie und Dystonie wurden ebenfalls beobachtet (Barkovich, 2002).
Holoprosenzephalie tritt in den ersten Wochen des intrauterinen Lebens auf. Die Prävalenz der Holoprosenzephalie in der frühen Embryonalentwicklung beträgt 1: 250 und sinkt auf 1:10.000-1: 20.000 zur Laufzeit (Nanni, 2000)., Es gibt keine bekannte Ursache für Holoprosenzephalie, obwohl viele Risikofaktoren vorgeschlagen wurden, einschließlich Diabetes bei Müttern (1% Risiko, 200-facher Anstieg) (Barra, 1983), Infektionen während der Schwangerschaft, wie TORCH-Infektionen (Munke, 1989) und Exposition gegenüber toxischen Substanzen, einschließlich Alkohol, Lithium, Thorazin, Hormone, Antikonvulsiva und Retinsäure (Nanni, 2000). Es wird davon ausgegangen, dass die meisten Fälle sporadisch auftreten, obwohl die Holoprosenzephalie auch eine genetische Grundlage hat., Familiäre Holoprosenzephalie wurde sowohl autosomal dominant als auch autosomal rezessiv vererbt gesehen. Chromosomenanomalien wurden auch mit Holoprosenzephalie in Verbindung gebracht, wobei Trisomie 13 die häufigste ist, obwohl dies keine konstante Assoziation ist (Kallen, 1992).Es wird angenommen, dass die Pathogenese einen Defekt in den Signalgenen beinhaltet, die für die Regulierung der Neuralrohrmuster verantwortlich sind., Intrakranielle Befunde umfassen eine variierende kortikale Hypoplasie, eine variierende Verschmelzung von Diencephalon, Basalganglien und Thalamus sowie das Vorhandensein einer dorsalen Zyste (aus verschmolzenem Thalami), die sich aus einem teilweise blockierten 3.Ventrikel ausdehnt (Simon, 2001). Hydrocephalus, verursacht durch abnormale Ansammlung von Liquor in den Ventrikeln, ist bei Holoprosenzephalie nicht ungewöhnlich und wird als auf Fehlbildung der Ventrikel oder übermäßige Liquorproduktion zurückzuführen angesehen., Dies erschwert oft die Holoprosenzephalie-Klassifikation, da das Gehirn komprimiert wird und sich der zuvor mikrozephale Schädel vor der Fusion der Schädelnähte ausdehnen kann (Tripathi, 2009). Es ist wichtig, die Vision des Patienten zu adressieren, um eine optimale Interaktion mit seiner Umgebung zu ermöglichen. Oft werden Gläser aufgrund des Grades der Gesichtsasymmetrie und der vorhandenen strukturellen Anomalien nicht toleriert. In diesen Situationen sind wir der Meinung, dass Kontaktlinsen als eine Möglichkeit zur Verbesserung der Sehfunktion in Betracht gezogen werden sollten.,
Obwohl es in der Allgemeinbevölkerung eine Variation der Insertion des medialen Rektusmuskels gibt, ist dieser Fall bemerkenswert für die anomalen extraokularen Muskeleinfügungen, insbesondere den 7 mm Abstand der medialen Recti vom Limbus. Wie oben erwähnt, sind Mittelliniendefekte bei der Holoprosenzephalie häufig, was erklären kann, warum die medialen Recti bevorzugt beteiligt waren. Mit Ausnahme der Zyklopie wurde wenig über okuläre und strabismische Assoziationen mit Holoprosenzephalie veröffentlicht.,
Insgesamt ist die Behandlung aufgrund der Schwere und Konfiguration der Fehlbildung des Patienten sehr individuell. Die Behandlung ist unterstützend und symptomatisch und die Prognose hängt stark von der Art der Holoprosenzephalie und den damit verbundenen Anomalien ab (Nanni, 2000).
In der Fall, die oben dargestellt, eine der wichtigsten überlegungen für den Patienten“die Familie war, wie man eingreifen, um vision zu verbessern und ermöglichen Sie wirtschaftliches Wachstum und Entwicklung. Der Patient war in den ersten Lebensjahren kritisch krank, und die Palliativversorgung wurde zunächst als praktikable Option diskutiert., Der patient“s-Familie wollte die Fortsetzung der Behandlung und versuchen, mögliche Interventionen zur Verbesserung der Lebensqualität. Angesichts solcher herausfordernder Situationen ist es für Gesundheitsdienstleister unerlässlich, Familien dabei zu unterstützen, Entscheidungen in einem respektvollen Umfeld ohne Urteilsvermögen oder unangemessenen Einfluss zu treffen. Das American College of Critical Care Medicine Task Force veröffentlicht Leitlinien für die klinische Praxis Adressierung Unterstützung der Familie in der patient-centered intensive care unit (Davidson, 2007)., Durch gute Kommunikations -, Konfliktmanagement-und Meeting-Moderationsfähigkeiten können Familien in ein gemeinsames Entscheidungsmodell eingebunden werden, in dem Familien nicht allein für alle medizinischen Entscheidungen autonom verantwortlich sind, noch medizinische Anbieter, die paternalistische Versorgung anbieten. Während Familientreffen wird empfohlen, Familienmitgliedern offene Fragen zu ihrem Verständnis der Versorgung des Patienten, seinen Ängsten und Bewältigungsstrategien zu stellen., Die Betreuer werden dann ermutigt, die Gefühle der Familie zu wiederholen, um das Vertrauen in das Team und den Entscheidungsprozess zu stärken. Danach sollten die Praktizierenden klare und ehrliche Informationen in zugänglicher Sprache bereitstellen und die Möglichkeit haben, Fragen zu stellen. Das Ziel der Diskussion ist der Konsens, der durch respektvolle Anerkennung aller Meinungen unterstützt wird.,
Penticuff und Arheart studierte die Wirksamkeit von Gesprächen zwischen dem Gesundheitsdienstleister und Eltern in einer Neugeborenen-Intensivstation Einstellung, und zeigte, dass „shared decision making“ führte zu weniger Konflikten, unrealistische elterliche Erwartungen, und verbessern die Zusammenarbeit, als auch Eltern helfen, besser zu verstehen, Ihr Kind die medizinische situation (Penticuff, 2005). Es wird gezeigt, dass der Stress in der Familie durch offene und effektive Kommunikation sowie ein Umfeld der Hoffnung verringert wird (Davidson, 2007)., Wichtig ist, dass eine solche patientenzentrierte Versorgung auch die klinischen Ergebnisse verbessert (Lewin, 2001).
Differentialdiagnose
- Esotropie
- Monokularer Höhenmangel
- Dissoziierte vertikale Abweichung
Zusammenfassung
|
Zeichen
|
Symptome
|
Behandlung
|
Barkovich AJ, Simon EM, Clegg NJ, Kinsman SL, Hahn JS. Analyse der Großhirnrinde bei Holoprosenzephalie mit Aufmerksamkeit auf die Sylvianfissuren. AJNR Am J Neuroradiol. 2002;23(1):143-50.
Blaas HG, Eriksson AG, Salvesen KA, Isaksen CV, Christensen B, Møllerløkken G, Eik-Nes SH., Gehirne und Gesichter bei Holoprosenzephalie: prä – und postnatale Beschreibung von 30 Fällen. Ultraschall Geburtshelfer Gynäkologe. 2002;19(1):24-38.
Barra Jr M, Hanson JW, Currey K, Scharf S, Toriello H, Schmickel RD, Wilson GA. Holoprosenzephalie bei Säuglingen diabetischer Mütter. J Pädiatrie 1983; 102: 565D8.
– Davidson JE, Powers K, Hedayat KM, Tieszen M, Kon AA, Shepard E, Spuhler V, Todres ID, Levy M, Barr J, Gandhi R, Hirsch G, Armstrong D., Richtlinien der klinischen Praxis zur Unterstützung der Familie auf der patientenzentrierten Intensivstation: American College of Critical Care Medicine Task Force 2004-2005. American College of Critical Care Medicine Task Force 2004-2005, Society of Critical Care Medicine. Crit Pflege Med. 2007;35(2):605-22.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007 2;2:8.
Kallen B, Castilla EE, Lancaster PAL, et al. Die Zyklopen und die Meerjungfrau: eine epidemiologische Studie über zwei Arten seltener Missbildungen. J Med Genet 1992;29:30-35.,
Lewin SA, Skea ZC, Entwistle V, Zwarenstein M, Dick J. Interventionen für Anbieter zur Förderung eines patientenzentrierten Ansatzes in klinischen Konsultationen. Cochrane Database Syst Rev. 2001;(4): CD003267. (PMID:11687181)
Munke M. Klinische, zytogenetische und molekulare Ansätze zur genetischen Heterogenität der Holoprosenzephalie. Am J Med Genet 1989;34:237-245.
Nanni L, Schelper RL, Muenke MT. Molekulargenetik der Holoprosenzephalie. Front Biosci. 2000 1;5:D334-42.
Penticuff JH, Arheart KL.,Wirksamkeit einer Intervention zur Verbesserung der elternprofessionellen Zusammenarbeit auf der Intensivstation von Neugeborenen. J Perinat Neonatal Nurs. 2005;19(2):187-202.
WIR Scott, Jackson OB. Double Elevator Lähmung: die Bedeutung der inferioren Rectus Einschränkung. Bin Orthopt J. 1977;27:5-10.
Simon EM, RF Hevner, Pinter J Clegg NJ, Delgado M, Verwandten SL, Hahn JS, Barkovich AJ. Die dorsale Zyste bei Holoprosenzephalie und die Rolle des Thalamus bei seiner Bildung. Neuroradiologie. 2001;43(9):787-91.
Tripathi AK, Agrawal D, Sedain G. Hydrocephalic holoprosencephaly: Ein oxymoron?, Einblicke in Ätiologie und Management. J Pädiatrische Neurowissenschaften. 2009;4(1):41-3.
Zustimmung zur Verwendung von Fotos und Videos von der Mutter des Patienten erhalten.
Vorgeschlagenes Zitierformat: Kemp PS, Casey, G, Longmuir SQ. Holoprosencephaly und Strabismus.Eyerounds.org. 12. Juni 2012, Verfügbar unter: http://EyeRounds.org/cases/151-holoprosencephaly-strabismus.htm