Holoprosencefalia y Estrabismo
Pavlina S. Kemp, MD, Grant Casey, Susannah Q. Longmuir, MD
12 de junio de 2012
queja principal: cruce de Ojos
historia de enfermedad presente
la paciente es una mujer de 15 meses de edad en el momento de la presentación en la clínica oftalmológica, con historia de hidrocefalia grave al nacer. También fue diagnosticada de holoprosencefalia alobar al nacer con convulsiones. Originalmente fue referida para cruzar los ojos. Presentamos su notable curso clínico.,
antecedentes médicos:
- hidrocefalia S/P derivación ventriculoperitoneal
- Holoprosencefalia (tipo alobar)
- trastorno convulsivo
antecedentes quirúrgicos:
- colocación de derivación Ventriculoperitoneal, 2004
- revisión de derivación Ventriculoperitoneal, 1/2005
- revisión de derivación ventriculoperitoneal, 6/2005
antecedentes familiares: no se conocen antecedentes familiares de holoprosencefalia, ambliopía o estrabismo.
Historia Social: la paciente vive en casa con sus padres y dos hermanas.,
medicamentos: ninguno
examen y Curso Clínico:
Edad: 15 meses
agudeza Visual: do Central, inestable y mantenida y SG central, inestable y mantenida
Prueba de agudeza del cajero:
- Sin corrección OU: 20/800
alumnos: igualmente redondos y defecto pupilar.,
Stereo Visión: incapaz de probar
motilidad y Estrabismo:
- Esotropia variable grande
- elevación Bilateral y déficit de abducción
- nistagmo horizontal intermitente
refracción Ciclopléjica:
- OD: +4.00
- OS: +6.00
examen externo: notable para la circunferencia de la cabeza grande
examen con lámpara de hendidura: Examen del segmento anterior normal ou sin evidencia de cataratas u otras opacidades de los medios.
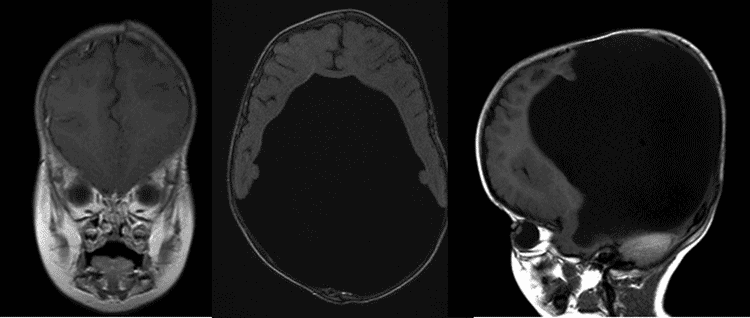
nervios ópticos de apariencia Normal y examen de fondo de ojo dilatado normal. No hay signos de hipoplasia del nervio óptico en ninguno de los ojos., Figura 1: resonancia magnética axial y sagital que ilustra el agrandamiento de los ventrículos secundario a hidrocefalia.
en este punto, después de la discusión con su familia, la cirugía para el estrabismo fue diferida y se intentó la corrección de su hipermetropía. Se recetaron anteojos. She was not able to wear glasses comfortably and contact lenses were tried. El parche se realizó para tratar su ambliopía. La paciente fue seguida y a la edad de 3 años, sus padres querían proceder con la cirugía de estrabismo para» destapar » sus ojos.,
Edad: 3
agudeza Visual: Central, do CUSM y OS CUSM
motilidad y Estrabismo:
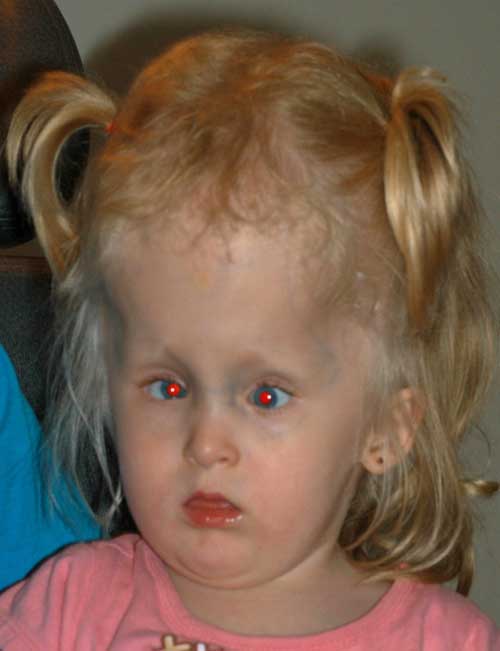
- esotropía de ángulo grande de 55-60 dioptrías prismáticas (Figura 2)
- déficit de abducción Bilateral y deficiencia de elevación monocular izquierda
en el momento de la cirugía de estrabismo, las ducciones forzadas intraoperatorias mostraron restricciones de ambos músculos del recto medial y se encontró que el paciente tenía una inserción anómala del músculo del recto medial. La inserción del músculo recto medial antes de la desinserción se encontró a 7 mm del limbo (más posterior de lo esperado a 5,5 mm del limbo). Secundario a que la anatomía no era normal, se realizó un abordaje conservador y se sometió a recesiones bilaterales del recto medial de 5,5 mm, dejando el recto medial en 12.,A 5 mm del limbus.
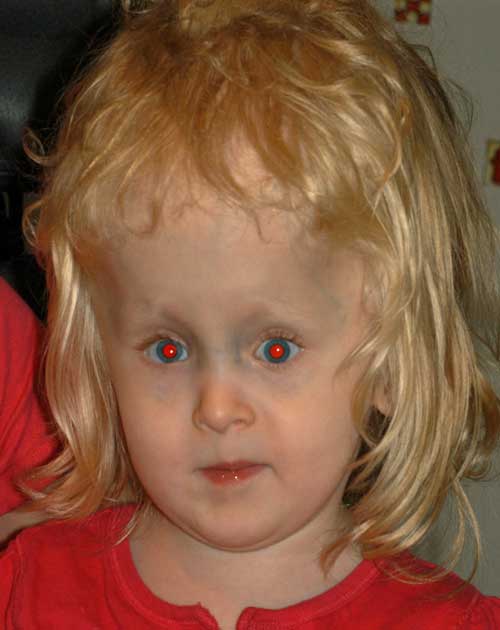
en el postoperatorio, tuvo una pequeña esotropía variable residual con pequeña desviación vertical y una deficiencia de elevación más notable del ojo izquierdo.Figura 3: fotos postoperatorias que demuestran una pequeña esotropía residual e hipotropía izquierda.
edad 6:
sus padres estaban contentos con la alineación; sin embargo, con el tiempo, desarrolló una hipertropia derecha notable. A los 6 años, su familia decidió proceder con una segunda cirugía de estrabismo para abordar la desalineación vertical.,
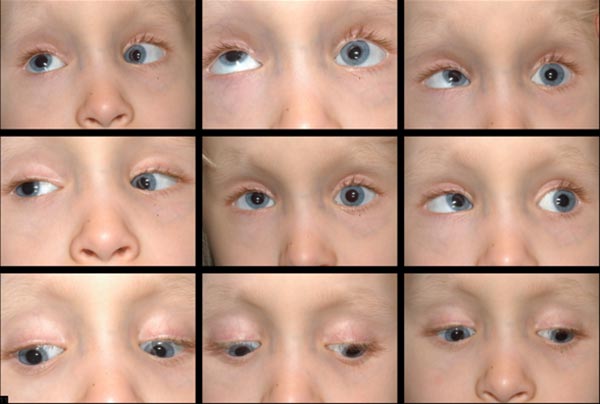
se planificó una recesión del recto inferior izquierdo vs una recesión del recto superior derecho, con decisión sobre qué procedimiento se realizaría con base en las ducciones forzadas intraoperatorias. Las ducciones forzadas intraoperatorias mostraron un recto inferior izquierdo apretado, lo que es consistente con la deficiencia de elevación monocular que habíamos considerado previamente. Se realizó una recesión del recto inferior izquierdo de 6 mm, desplazando el músculo de 8 mm posterior al limbo, donde fue encontrado, a 14 mm posterior al limbo.,Figura 5: foto postoperatoria que muestra una pequeña esotropía residual e hipotropía izquierda (~7δ).
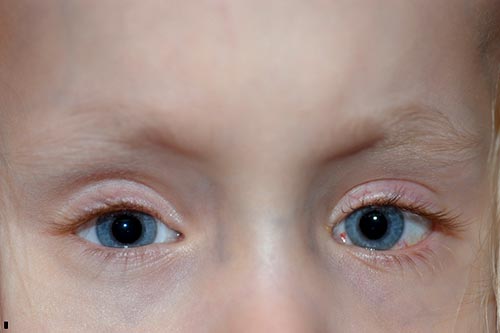
unos meses después de su segunda cirugía, su madre comenzó a notar que su ojo derecho se desplazaba hacia arriba en momentos de falta de atención. Fue vista en la clínica y notó que tenía una desviación vertical disociada manifiesta a la derecha., Se decidió proceder de nuevo con la cirugía de estrabismo y una recesión del recto superior derecho de 8 mm por el método de hangback, moviendo el músculo de 8 mm posterior al limbo, donde fue encontrado, a 16 mm posterior al limbo.
la paciente tuvo un buen desempeño postoperatorio (Figura 6), y ha ido desarrollando progresivamente sus habilidades visuales. Lo ha hecho muy bien en general y está caminando y ahora leyendo. figura 6: foto postoperatoria que muestra una alineación ocular satisfactoria.,
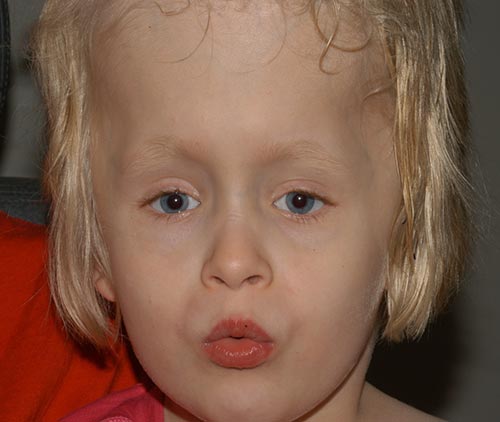
diagnóstico:
Holoprosencefalia con esotropía infantil y estrabismo complejo, con variabilidad en las inserciones musculares del recto medial.
discusión:
La Holoprosencefalia es un tipo de trastorno cefálico caracterizado por el fracaso del prosencéfalo (el cerebro anterior embrionario) para desarrollarse, lo que lleva a una estructura cerebral de un solo lóbulo y graves defectos craneales y faciales., Hay tres clasificaciones de holoprosencefalia: holoprosencefalia alobar, holoprosencefalia semi-lobar y holoprosencefalia lobar.
la holoprosencefalia de Alobar representa dos tercios de los pacientes afectados, y es la forma más grave, caracterizada por el fracaso del cerebro para separarse en dos mitades. Esto resulta en un solo ventrículo primitivo, ausencia de bulbos olfativos y tractos ópticos y anomalías graves del desarrollo. Por lo general, se asocia con anomalías faciales graves, que incluyen ojos muy espaciados, cabeza pequeña, labio leporino y paladar hendido., La holoprosencefalia semilobar, que representa una cuarta parte de los casos de holoprosencefalia, es una forma intermedia de la enfermedad y se caracteriza por hemisferios cerebrales parcialmente separados y un solo ventrículo. La holoprosencefalia Lobar es la forma menos grave, en la que el cerebro del paciente puede ser casi normal; hay una fisura distinta entre los lóbulos centrales desarrollados, y está presente alguna fusión de las estructuras cerebrales (Nanni, 2000). En la mayoría de los casos de holoprosencefalia, las malformaciones cerebrales son incompatibles con la vida., En los casos menos graves, los bebés nacen con un desarrollo cerebral normal o casi normal y diversos grados de deformidad facial.
Los defectos craneofaciales de la línea media son el sello distintivo de la holoprosencefalia y pueden incluir microcefalia, hipotelorismo (ojos anormalmente espaciados), anomalías nasales, como aplanamiento nasal o un solo naris, y los defectos del labio superior y del paladar, como paladar hendido o un solo incisivo frontal. La ciclopía puede estar presente en las formas más severas donde una probóscide similar a la nariz está presente sobre un solo ojo en el Centro de la cara (Nanni, 2000)., Se cree que el grado de deformidad facial indica la gravedad de los defectos intracraneales. Las comorbilidades asociadas incluyen disfunción de la glándula pituitaria y el hipotálamo, lo que resulta en desregulación de la temperatura corporal, convulsiones y retraso mental de diversa gravedad (Dubourg, 2007). También se han observado hipotonía y distonía (Barkovich, 2002).
La Holoprosencefalia ocurre durante las primeras semanas de vida intrauterina. La prevalencia de la holoprosencefalia en el desarrollo embrionario temprano es de 1:250, disminuyendo a 1:10.000-1:20.000 a término (Nanni, 2000)., No se conoce la causa de la holoprosencefalia, aunque se han sugerido muchos factores de riesgo, incluida la diabetes materna (riesgo del 1%, aumento de 200 veces) (Barra, 1983), infecciones durante el embarazo, como las infecciones por TORCH (Munke, 1989), y la exposición a sustancias tóxicas, como alcohol, litio, Torazina, hormonas, anticonvulsivos y ácido retinoico (Nanni, 2000). Se considera que la mayoría de los casos ocurren esporádicamente, aunque se ha encontrado que la holoprosencefalia también tiene una base genética., La holoprosencefalia familiar se ha visto heredada tanto en patrones autosómicos dominantes como autosómicos recesivos. Las anomalías cromosómicas también se han asociado con holoprosencefalia, siendo la trisomía 13 la más común, aunque esta no es una asociación constante (Kallen, 1992).
Se cree que la patogénesis implica un defecto en los genes de señalización responsables de regular el patrón del tubo neural., Los hallazgos intracraneales incluyen hipoplasia cortical variable, fusión variable del diencéfalo, ganglios basales y tálamo y presencia de un quiste dorsal (que surge de tálamo fusionado) que se expande a partir de un 3er ventrículo parcialmente bloqueado (Simon, 2001). La hidrocefalia, causada por una acumulación anormal de LCR en los ventrículos, no es infrecuente en la holoprosencefalia y se cree que se debe a una malformación de los ventrículos o a una producción excesiva de LCR., Esto a menudo complica la clasificación de la holoprosencefalia, ya que el cerebro se comprime y se permite que el cráneo previamente microcefálico se expanda antes de la fusión de las suturas craneales (Tripathi, 2009). Es importante abordar la visión del paciente para permitir una interacción óptima con su entorno circundante. A menudo, las gafas no se toleran debido al grado de asimetría facial y las anomalías estructurales presentes. En estas situaciones, creemos que las lentes de contacto deben considerarse como una forma de mejorar la función visual.,
aunque hay variación de la inserción muscular del recto medial en la población general, este caso es notable por las inserciones musculares extraoculares anómalas, específicamente la distancia de 7 mm del recto medial desde el limbo. Como se mencionó anteriormente, los defectos de la línea media son comunes en la holoprosencefalia, lo que puede explicar por qué los rectos mediales estaban involucrados preferentemente. Con la excepción de ciclopía, poco se ha publicado sobre el ocular y estrábico asociaciones con holoprosencefalia.,
En general, el tratamiento es altamente individualizado en función de la gravedad del paciente y la configuración de la malformación. El tratamiento es sintomático y de apoyo y el pronóstico depende en gran medida del tipo de holoprosencefalia y sus anomalías asociadas (Nanni, 2000).
en el caso presentado anteriormente, una de las consideraciones más significativas para la familia del paciente fue cómo intervenir para mejorar la visión y permitir un crecimiento y desarrollo continuos. El paciente estaba críticamente enfermo en los primeros años de vida, y los cuidados paliativos se discutieron inicialmente como una opción viable., La familia del paciente quería continuar el tratamiento y buscar posibles intervenciones para mejorar la calidad de vida. Cuando se enfrentan a situaciones desafiantes como esta, es esencial que los proveedores de atención médica ayuden a las familias a tomar decisiones en un ambiente respetuoso sin juicios ni influencias indebidas. El American College of Critical Care Medicine Task Force publicó guías de práctica clínica que abordan el apoyo de la familia en la unidad de cuidados intensivos centrada en el paciente (Davidson, 2007)., A través de una buena comunicación, manejo de conflictos y habilidades de facilitación de reuniones, las familias pueden participar en un modelo compartido de toma de decisiones en el que las familias no son las únicas responsables de todas las decisiones médicas de forma autónoma, ni los proveedores médicos proporcionan atención paternalista. Durante las reuniones familiares, se recomienda que a los miembros de la familia se les hagan preguntas abiertas sobre su comprensión del cuidado del paciente, sus miedos y estrategias de afrontamiento., Luego, se anima a los proveedores de atención a repetir los sentimientos de la familia para permitir el desarrollo de la confianza en el equipo y en el proceso de toma de decisiones. Después de esto, los profesionales deben proporcionar información clara y honesta en un lenguaje accesible, con la oportunidad de hacer preguntas. El objetivo de la discusión es el consenso, que es ayudado por el reconocimiento respetuoso de todas las opiniones.,
Penticuff y Arheart estudiaron la efectividad de las reuniones entre proveedores de atención médica y padres en un entorno de unidad de cuidados intensivos neonatales, y mostraron que la toma de decisiones compartida resultó en menos conflictos, expectativas poco realistas de los padres y una mejor colaboración, además de ayudar a los padres a comprender mejor la situación médica de su hijo (Penticuff, 2005). Los niveles de estrés familiar se muestran disminuidos con una comunicación abierta y efectiva, así como con un ambiente de esperanza (Davidson, 2007)., Es importante destacar que este tipo de atención centrada en el paciente también ha demostrado mejorar los resultados clínicos (Lewin, 2001).
diagnóstico diferencial
- esotropia
- deficiencia de elevación Monocular
- desviación vertical disociada
resumen
|
signos
|
síntomas
|
tratamiento
|
barkovich aj, Simon em, Clegg NJ, Kinsman SL, Hahn JS. Análisis de la corteza cerebral en holoprosencefalia con atención a las fisuras sylvianas. AJNR Am J Neuroradiol. 2002;23(1):143-50.
Blaas HG, Eriksson AG, Salvesen KA, Isaksen CV, Christensen B, Møllerløkken G, Eik-Nes SH., Brains and faces in holoprosencephaly: pre – and postnatal description of 30 cases. Ultrasonido Obstet Ginecol. 2002;19(1):24-38.Barra Jr M, Hanson JW, Currey K, Sharp s, Toriello H, Schmickel RD, Wilson GA. Holoprosencefalia en bebés de madres diabéticas. J Pediatr 1983; 102: 565D8.Davidson JE, Powers K, Hedayat KM, Tieszen M, Kon AA, Shepard e, Spuhler V, Todres ID, Levy M, Barr J, Ghandi R, Hirsch G, Armstrong D., Clinical practice guidelines for support of the family in the patient-centered intensive care unit: American College of Critical Care Medicine Task Force 2004-2005. American College of Critical Care Medicine Task Force 2004-2005, Society of Critical Care Medicine. Crit Care Med. 2007;35(2):605-22.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent s, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007 2;2:8.
Kallen B, Castilla EE, Lancaster PAL, et al. The cyclops and the mermaid: an epidemiological study of two types of rare malformation (en inglés). J Med Genet 1992; 29: 30-35.,
Lewin SA, Skea ZC, Entwistle V, Zwarenstein M, Dick J. Interventions for providers to promote a patient-centred approach in clinical consultations. Cochrane Database Syst Rev. 2001; (4): CD003267. (PMID:11687181)
Munke M. Clinical, citogenetic and molecular approaches to the genetic heterogeneity of holoprosencephaly. Am J Med Genet 1989; 34: 237-245.
Nanni L, Schelper RL, Muenke MT. Molecular genetics of holoprosencephaly. Biosci Frontal. 2000 1; 5: D334-42.
Penticuff JH, Arheart KL.,Eficacia de una intervención para mejorar la colaboración entre padres y profesionales en Cuidados Intensivos Neonatales. J Perinat Neonatal Nurs. 2005;19(2):187-202.Scott WE, Jackson OB. Parálisis de doble Ascensor: la importancia de la restricción del recto inferior. Am Orthopt J. 1977; 27: 5-10.
Simon EM, Hevner RF, Pinter J, Clegg NJ, Delgado M, Kinsman SL, Hahn JS, Barkovich AJ. El quiste dorsal en la holoprosencefalia y el papel del tálamo en su formación. Neurorradiología. 2001;43(9):787-91.
Tripathi AK, Agrawal D, Sedain G. Hydrocephalic holoprosencephaly: An oxymoron?, Insights into etiology and management (en inglés). J Pediatr Neurosci. 2009;4(1):41-3.
consentimiento para el uso de fotos y videos obtenidos de la madre del paciente.
Formato de cita sugerido: Kemp PS, Casey, G, Longmuir SQ. Holoprosencefalia y Strabismus.Eyerounds.org. 12 de junio de 2012, Disponible en: http://EyeRounds.org/cases/151-holoprosencephaly-strabismus.htm