Was sind die Unterschiede in den FDA-Medizinprodukteklassen?
Die US FDA regelt alle in den USA vertriebenen Medizinprodukte, die in drei große Klassen eingeteilt sind. Jedes von der FDA zugelassene Medizinprodukt wird je nach Risiko, Invasivität und Auswirkungen auf die allgemeine Gesundheit des Patienten entweder als Klasse I, II oder III eingestuft. Aber wo sind die Linien zwischen jeder dieser drei Klassen gezogen, und warum?,
Die Klassifizierungsrichtlinien der US FDA können für Hersteller von Medizinprodukten mit begrenzter Exposition gegenüber dem System sehr verwirrend sein. Je nachdem, wie Ihr Gerät gruppiert ist, gibt es einen enormen Unterschied im optimalen Marktweg für Hersteller. Geräte der Klasse I unterliegen weitaus weniger regulatorischen Anforderungen als Geräte der Klasse II oder III.
Wenn Sie die Unterschiede in den FDA – Medizinprodukteklassen verstehen, können Sie verstehen, wie Ihr Gerät gruppiert wird., Mit diesem Wissen können Medizinproduktehersteller in den Vormarktphasen die für die behördliche Genehmigung erforderlichen Ressourcen besser vorbereiten und zuweisen.
Unterschiede zwischen FDA-Medizinproduktklassen
Die FDA hat über 1.700 verschiedene Arten von Medizinprodukten klassifiziert. Die Geräte sind im Code of Federal Regulations (CFR) nach 16 Fachgebieten wie kardiovaskulären oder hämatologischen Geräten organisiert., Die Klassifizierung Ihres Medizinprodukts nach einem der 16 Fachgebiete ist der erste Schritt, um zu verstehen, ob Sie ein Medizinprodukt der Klassen I, II oder III herstellen.
Nach der Klassifizierung eines Geräts nach Spezialität weist die FDA die Hersteller an, mit der Vorabbenachrichtigung fortzufahren, wenn sie wissen, ob ihr Gerät befreit ist oder nicht. Medizinprodukte der Klasse I, die am wenigsten riskante und invasive Kategorie, sind von Vorabbenachrichtigungsprozessen ausgenommen. Bestimmte Geräte der Klasse II sind ebenfalls von der Vorkaufsgenehmigung ausgenommen.,
Alle von der FDA regulierten Geräte unterliegen jedoch den aktuellen Anforderungen der Good Manufacturing Practice (cGMP) an Registrierung, Kennzeichnung und Qualität. Aber woher wissen Sie, ob Ihr Gerät Klasse I oder II ist und ob Sie sich einer Vorverkaufsbenachrichtigung unterziehen müssen? 
Class 1
Die US FDA definiert Geräte der Klasse I als Geräte, die “ nicht zur Unterstützung oder Aufrechterhaltung des Lebens oder von wesentlicher Bedeutung zur Verhinderung von Beeinträchtigungen der menschlichen Gesundheit bestimmt sind und möglicherweise kein potenzielles unangemessenes Risiko für Krankheiten oder Verletzungen darstellen.,“
Diese Geräte sind die häufigste Klasse von Geräten, die von der FDA reguliert werden und 47 Prozent der auf dem Markt zugelassenen Geräte ausmachen.
Geräte der Klasse I haben einen minimalen Kontakt mit Patienten und geringe Auswirkungen auf die allgemeine Gesundheit eines Patienten. Im Allgemeinen kommen Geräte der Klasse I nicht mit den inneren Organen eines Patienten, dem Zentralnervensystem oder dem Herz-Kreislauf-System in Kontakt. Diese Geräte unterliegen den wenigsten gesetzlichen Anforderungen.,
Beispiele für Geräte der Klasse I:
- Elektrische Zahnbürste
- Zungendepressor
- Sauerstoffmaske
- Wiederverwendbares chirurgisches Skalpell
- Bandagen
- Krankenhausbetten
Medizinische Geräte der Klasse I auf den Markt bringen
Geräte der Klasse I sind am schnellsten und am einfachsten auf den Markt zu bringen, da sie das geringste Risiko für den Patienten darstellen und selten entscheidend für die lebenserhaltende Pflege. Die meisten Geräte der Klasse I sind von den FDA-Anforderungen für Premarket Notification (510k) und Premarket Approval (PMA) ausgenommen.,
Geräte der Klasse I sind nicht von den allgemeinen FDA-Kontrollen befreit, einer Reihe von Befehlen, die für medizinische Geräte der Klassen I, II und III gelten. Die Bestimmungen dieses Gesetzes befassen sich mit Verfälschung, Markenmissbrauch, Geräteregistrierung, Aufzeichnungen und guten Herstellungspraktiken. Hersteller von Medizinprodukten, die in die Klasse A fallen, müssen weiterhin ein Qualitätsmanagementsystem implementieren und Standards einhalten, um ein Qualitätsprodukt sicherzustellen.,
VERWANDTE LESEN: Der Unterschied Zwischen Vorbörslich Notification 510(k) – und Vorbörslich Genehmigung
Class 2
Klasse II medizinische Geräte sind komplizierter als Klasse-I-Geräte und präsentieren Sie in einer höheren Kategorie Risiko, weil Sie mehr sind wahrscheinlich zu kommen, in längeren Kontakt mit einem Patienten. Dies kann Geräte umfassen, die mit dem Herz-Kreislauf-System oder den inneren Organen eines Patienten in Kontakt kommen, und diagnostische Werkzeuge.,
Die FDA definiert Geräte der Klasse II als “ Geräte, für die allgemeine Kontrollen nicht ausreichen, um die Sicherheit und Wirksamkeit des Geräts angemessen zu gewährleisten.,est Kits
Bringen Klasse II Medizinische Geräte zu Markt
Kontrollen variieren je nach gerät, aber nach der FDA, können umfassen:
- Gerät leistung
- Postmarket überwachung
- Patienten register
- Spezielle Kennzeichnungsanforderungen
- Vorkaufsdatenanforderungen
- Richtlinien
li>
Die Mehrheit der Klasse-II-Geräte ist FDA-zugelassen für den Markt durch die Vorkaufsanzeige oder 510(k) Prozess.,
Geräte der Klasse II unterliegen denselben allgemeinen Kontrollen, die oben erwähnt wurden, aber die FDA definiert sie als „Geräte, für die allgemeine Kontrollen nicht ausreichen, um die Sicherheit und Wirksamkeit des Geräts angemessen zu gewährleisten.“Aus diesem Grund unterliegen Geräte der Klasse II auch speziellen Kontrollen. Diese Vorschriften hängen vom Gerät ab und können spezielle Kennzeichnungsanforderungen, Patientenregistrierungen und Leistungsstandards enthalten.
Die meisten Geräte der Klasse II kommen mit dem Premarket Notification (510k) – Verfahren auf den Markt., Die 510 (k) ist eine komplexe Anwendung bei der FDA, die zeigt, dass ein Gerät sicher und effektiv ist, indem sie demonstriert, dass das Gerät einem anderen Gerät entspricht, das auf dem Markt ist.
Bei diesem Vorgang wird eine „substantielle Äquivalenz“ zu einem anderen Gerät angezeigt, das im amerikanischen Sprachgebrauch als „Prädikat“ bekannt ist.“Dies bedeutet nicht, dass die Geräte identisch sein müssen, aber sie erfordern erhebliche Ähnlichkeiten in der Verwendung, Design, Materialien, Kennzeichnung, Standards und andere Eigenschaften.,
Die FDA hat Anfang 2018 eine Freistellungsliste veröffentlicht, die über 800 generische Medizinprodukte der Klasse I und II vom 510(k) – Prozess befreit. Wenn Sie über ein generisches Medizinprodukt der Klasse II verfügen, können Sie anhand der FDA-Produktklassifizierungsdatenbank feststellen, ob es von einer 510(k) – Anmeldung befreit ist.
VERWANDTE LESEN: 5 Gründe, die zur Überholung von der FDA 510(k) ein Toller Zug.
Klasse 3
Die FDA definiert Geräte der Klasse III als Produkte, die „in der Regel das Leben erhalten oder unterstützen, implantiert werden oder ein potenzielles unangemessenes Risiko für Krankheiten oder Verletzungen darstellen.,“
Nur 10 Prozent der von der US FDA regulierten Geräte fallen in die Klasse III. Diese Klassifizierung wird im Allgemeinen auf permanente Implantate, intelligente medizinische Geräte und Lebenserhaltungssysteme ausgedehnt.
Während die Klasse III im Allgemeinen den innovativsten und modernsten medizinischen Geräten vorbehalten ist, gibt es andere Geräte, die aus verschiedenen Gründen in die Klasse III fallen können., Einige Geräte, die zunächst als Klasse II eingestuft werden, können auf Klasse III angehoben werden, wenn der Hersteller während des PMA-Anmeldeverfahrens (510k) keine wesentliche Äquivalenz zu einem Prädikat (bestehendes Produkt) nachweisen kann.,
Beispiele für medizinische Geräte der Klasse III:
- Brustimplantate
- Herzschrittmacher
- Defibrillatoren
- Hochfrequenzventilatoren
- Cochlea-Implantate
- Fötale Blutentnahmemonitore
- Implantierte Prothetik
Medizinische Geräte der Klasse III auf den Markt bringen
Geräte der Klasse III unterliegen der FDA Allgemeine Kontrollen und der FDA Premarket Approval (PMA) Prozess., Die FDA schreibt: „Aufgrund des mit Geräten der Klasse III verbundenen Risikos hat die FDA festgestellt, dass allgemeine und spezielle Kontrollen allein nicht ausreichen, um die Sicherheit und Wirksamkeit von Geräten der Klasse III zu gewährleisten.“
Die PMA ist die intensivste Art von Device-Marketing-Anwendung, die von der FDA benötigt wird. Einige FDA-Klasse-III-Geräte sind ausgenommen und können für eine 510(k) Einreichung qualifizieren, aber die Mehrheit wird erwartet, dass Premarket-Zulassung zu gewinnen.,
Der PMA-Prozess erfordert eine strenge Untersuchung eines Medizinprodukts, um Sicherheit und Wirksamkeit durch die Entwicklung eines datengetriebenen Nutzen-Risiko-Profils nachzuweisen. Der PMA-Prozess umfasst im Allgemeinen klinische Studien und erhebliche Zeit und Ressourcen für eine ausreichende Datenerfassung. Die einzigen Ausnahmen vom PMA-Verfahren innerhalb der Klasse III sind Geräte mit einem wesentlichen Äquivalent. Sie können bestimmen, ob ein Gerät der Klasse III mit einem 510(k) vermarktet werden kann, indem Sie die PMA-Datenbank (FDA Premarket Approval) und die 510(k) Premarket Notification-Datenbank durchsuchen.,
So bestimmen Sie Ihre Klasse
Der erste Schritt zur Klassifizierung Ihres Medizinprodukts besteht darin, durch die FDA-Klassifizierungsvorschriften zu navigieren, die Liste der 16 Kategorien für Medizinprodukte gemäß der medizinischen Spezialisierung.
Als Beispiel zeigen wir Ihnen die Schritte zur Identifizierung der Klassifizierung eines Blutdruckalarms. Das Gerät ist in die Kategorie 870 eingestuft: Herz-Kreislauf-Geräte.,
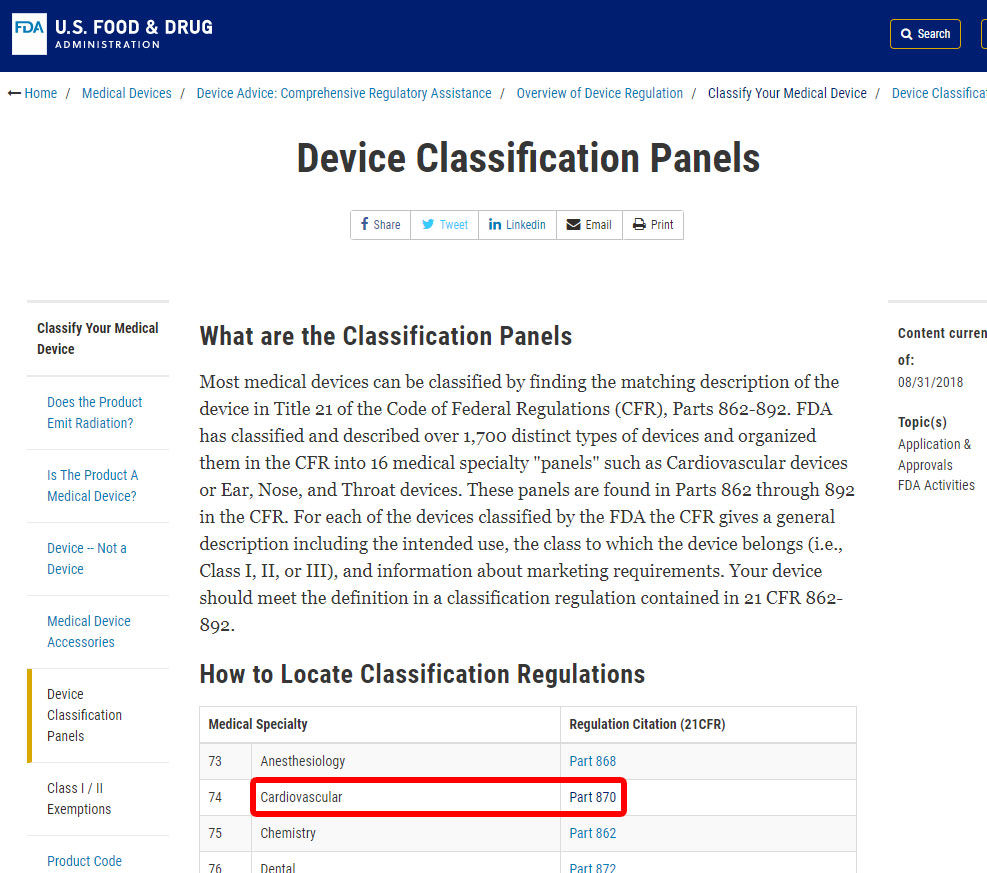
Sobald Sie die entsprechende medizinische Fachrichtung gefunden haben, klicken Sie auf die Kategorie und navigieren Sie durch die Liste der Geräte, bis Sie ein Äquivalent und den zugehörigen Gerätecode finden.
Klicken Sie auf den Gerätecode und öffnen Sie die Richtlinien. Die Geräteklassifikation ist unter Abschnitt (b) aufgeführt.,
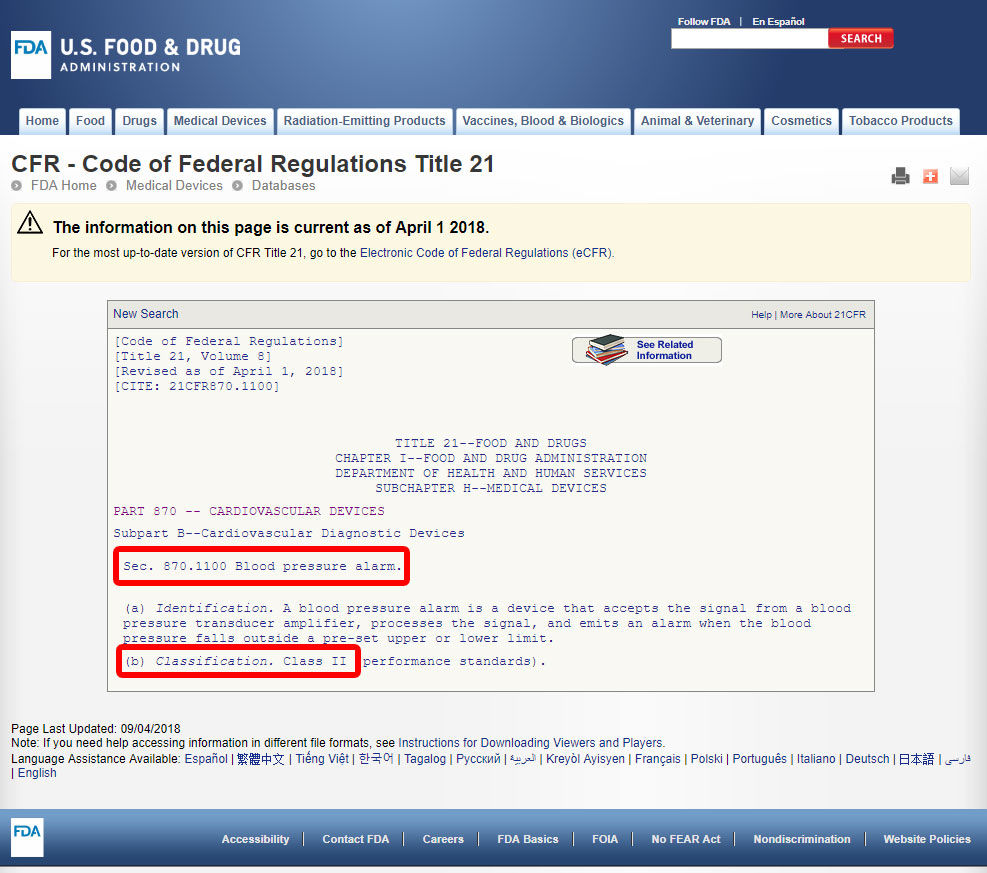
Wenn Ihrem Gerät ein aufgelistetes Äquivalent unter den 1.700 von der FDA klassifizierten Geräten fehlt, handelt es sich höchstwahrscheinlich um ein innovatives Gerät ohne ein wesentliches Äquivalent und würde als Klasse III klassifiziert.
Verständnis der FDA-Medizinproduktklassen
Die Unterschiede zwischen Medizinprodukten, die von der FDA als Klasse I, II oder III klassifiziert werden, sind hauptsächlich Risiko, Menge des Kontakts mit einem Patienten und seinen internen Systemen und ob ein Gerät das Leben erhalten.,
Zusätzlich zu diesen Faktoren berücksichtigt die FDA eine erhebliche Äquivalenz bei der Bestimmung der Klassifizierung eines Geräts. Wenn Ihr Gerät ein geringes Risiko aufweist und den Patienten minimal kontaktiert, qualifizieren Sie sich wahrscheinlich für Klasse I und einen optimierten Marktzulassungsprozess. Geräte der Klassen II und III müssen Sicherheit durch materielle Äquivalenz, eine 510(k) – Einreichung oder das Vorabgenehmigungsverfahren nachweisen.
Wenn Sie wissen, wie Ihr Gerät klassifiziert ist, können Sie Ihren Weg zur Marktzulassung rationalisieren, indem Sie die Prozesse und Dokumente verstehen, die wahrscheinlich von der FDA benötigt werden., Wenn Ihre Organisation einer Klasse-II-oder Klasse-III-Klasse 510(k) oder PMA-Anforderung unterliegt, können Sie mit diesem Wissen die entsprechenden Ressourcen im Voraus zuweisen und eine erfolgreiche Einreichung planen.
