¿Cuáles son las diferencias en las clases de dispositivos médicos de la FDA?
la FDA de los Estados Unidos regula todos los dispositivos médicos comercializados en los Estados Unidos, que se agrupan en tres clases generales. Cualquier dispositivo médico aprobado por la FDA se clasifica como clase I, II o III dependiendo del riesgo del dispositivo, la invasividad y el impacto en la salud general del paciente. Pero, ¿dónde están las líneas trazadas entre cada una de estas tres clases, y por qué?,
las pautas de clasificación de la FDA de los Estados Unidos pueden ser muy confusas para los fabricantes de dispositivos médicos con exposición limitada al sistema. Hay una enorme diferencia en el camino óptimo al mercado para los fabricantes dependiendo de cómo se agrupe su dispositivo. Los dispositivos de clase I están sujetos a muchos menos requisitos reglamentarios que los dispositivos de clase II o III.
al comprender las diferencias en las clases de dispositivos médicos de la FDA, puede comprender cómo se agrupará su dispositivo., Con este conocimiento en la mano, los fabricantes de dispositivos médicos en las etapas previas a la comercialización pueden preparar y asignar mejor los recursos necesarios para la aprobación regulatoria.
diferencias entre las clases de dispositivos médicos de la FDA
la FDA ha clasificado más de 1,700 tipos distintos de dispositivos médicos. Los dispositivos están organizados en el código de Regulaciones Federales (CFR) de acuerdo con 16 especialidades, como los dispositivos cardiovasculares o hematológicos., Clasificar su dispositivo médico de acuerdo con una de las 16 especialidades es el primer paso para comprender si está fabricando un dispositivo médico de clase I, II o III.
después de clasificar un dispositivo de acuerdo con la especialidad, la FDA instruye a los fabricantes a proceder a la notificación previa a la comercialización con el conocimiento de si su dispositivo está exento o no. Los dispositivos médicos de clase I, la categoría menos riesgosa e invasiva, están exentos de los procesos de notificación previa a la comercialización. Los dispositivos específicos de clase II también están exentos de la aprobación previa a la comercialización.,
sin embargo, todos los dispositivos regulados por la FDA están sujetos a los requisitos actuales de Buenas Prácticas de fabricación (cGMP) para el registro, el etiquetado y la calidad. Pero, ¿cómo sabes si tu dispositivo es de clase I O II, y si tienes que someterte a una notificación previa a la comercialización? 
Clase 1
la FDA de los EE.UU. define los dispositivos de clase I como los dispositivos que «no están destinados a ser utilizados para apoyar o sostener la vida o de importancia sustancial para prevenir el deterioro de la salud humana, y no pueden presentar un riesgo potencial irrazonable de enfermedad o lesión.,»
estos dispositivos son la clase más común de dispositivos regulados por la FDA, constituyendo el 47 por ciento de los dispositivos aprobados en el mercado.
los dispositivos de clase I tienen un contacto mínimo con los pacientes y un bajo impacto en la salud general del paciente. En general, los dispositivos de clase I no entran en contacto con los órganos internos del paciente, el sistema nervioso central o el sistema cardiovascular. Estos dispositivos están sujetos al menor número de requisitos reglamentarios.,
ejemplos de dispositivos de clase I:
- Cepillo de Dientes Eléctrico
- depresor de lengua
- Máscara de oxígeno
- bisturí quirúrgico reutilizable
- vendajes
- camas de Hospital
llevar al mercado Dispositivos Médicos de clase I
Los dispositivos de clase I son los más rápidos y fáciles de llevar al mercado, ya que presentan la menor cantidad de riesgo para el paciente y atención para el mantenimiento de la vida. La mayoría de los dispositivos de clase I están exentos de los requisitos de la FDA para la notificación previa a la comercialización (510k) y la aprobación previa a la comercialización (PMA).,
los dispositivos de clase I no están exentos de los controles generales de la FDA, una serie de órdenes que se aplican a los dispositivos médicos de clase I, II y III. Las disposiciones de esta ley se refieren a la adulteración, las marcas incorrectas, el registro de dispositivos, los registros y las buenas prácticas de fabricación. Los fabricantes de dispositivos médicos que entran en la clase A todavía están obligados a implementar un sistema de gestión de calidad y seguir los estándares para garantizar un producto de calidad.,
lectura relacionada: la diferencia entre la notificación previa a la comercialización 510(k) y la aprobación previa a la comercialización
clase 2
los dispositivos médicos de clase II son más complicados que los dispositivos de clase I y presentan una categoría de riesgo más alta porque tienen más probabilidades de entrar en contacto sostenido con un paciente. Esto puede incluir dispositivos que entran en contacto con el sistema cardiovascular o los órganos internos del paciente, y herramientas de diagnóstico.,
la FDA define los dispositivos de clase II como «dispositivos para los cuales los controles generales son insuficientes para proporcionar una garantía razonable de la seguridad y eficacia del dispositivo.,Kits est
llevar al mercado Dispositivos Médicos de clase II
Los controles varían según el dispositivo, pero según la FDA, pueden incluir:
- rendimiento del dispositivo
- vigilancia postcomercialización
- registros de pacientes
- requisitos especiales de etiquetado
- requisitos de datos previos a la comercialización
- pautas
La mayoría de los dispositivos de clase II están aprobados por la FDA para el mercado a través del proceso de notificación previa a la comercialización o 510(K).,
los dispositivos de clase II están sujetos a los mismos controles generales mencionados anteriormente, pero la FDA los define como » dispositivos para los cuales los controles generales son insuficientes para proporcionar una garantía razonable de la seguridad y eficacia del dispositivo.»Por esa razón, los dispositivos de clase II también están sujetos a controles especiales. Estas regulaciones dependen del dispositivo y pueden incluir requisitos especiales de etiquetado, registros de pacientes y estándares de rendimiento.
La mayoría de los dispositivos de clase II llegan al mercado utilizando el proceso de notificación previa al mercado (510k)., El 510 (k) es una aplicación compleja para la FDA, que demuestra que un dispositivo es Seguro y efectivo al demostrar que el dispositivo es equivalente a otro dispositivo que está en el mercado.
este proceso implica mostrar » equivalencia sustancial «a otro dispositivo que se conoce en el lenguaje de la FDA como» el predicado.»Esto no significa que los dispositivos deban ser idénticos, pero requieren similitudes significativas en el uso, diseño, materiales, etiquetado, estándares y otras características.,
la FDA publicó una lista de exenciones a principios de 2018 que exime a más de 800 dispositivos médicos genéricos de clase I y II del proceso 510(k). Si tiene un dispositivo médico genérico de clase II, puede descubrir si está exento de una presentación 510(k) buscando en la base de datos de clasificación de productos de la FDA.
lectura relacionada: 5 Razones por las que revisar la FDA 510 (k) es un gran paso.
Clase 3
la FDA define los dispositivos de clase III como productos que «por lo general sostienen o sostienen la vida, se implantan o presentan un riesgo potencial irrazonable de enfermedad o lesión.,»
solo el 10 por ciento de los dispositivos regulados por la FDA de los Estados Unidos entran en la clase III. esta clasificación generalmente se extiende a los implantes permanentes, los dispositivos médicos inteligentes y los sistemas de soporte vital.
mientras que la clase III generalmente se reserva para los dispositivos médicos más innovadores y de vanguardia, hay otros dispositivos que pueden caer en la clase III por diferentes razones., Algunos dispositivos que se clasifican inicialmente como clase II pueden ser ascendidos a clase III si el fabricante no puede demostrar una equivalencia sustancial con un predicado (producto existente) durante el proceso de presentación PMA (510k).,
ejemplos de Dispositivos Médicos de clase III:
- implantes mamarios
- marcapasos
- desfibriladores
- ventiladores de alta frecuencia
- implantes cocleares
- monitores de muestreo de sangre Fetal
- prótesis implantadas
llevar al mercado Dispositivos Médicos de clase III
controles generales y el proceso de aprobación previa a la comercialización (PMA) de la FDA., La FDA escribe: «debido al nivel de riesgo asociado con los dispositivos de clase III, LA FDA ha determinado que los controles generales y especiales por sí solos son insuficientes para garantizar la seguridad y eficacia de los dispositivos de clase III.»
el PMA es el tipo más intensivo de aplicación de marketing de dispositivos requerido por la FDA. Algunos dispositivos de clase III de la FDA están exentos y pueden calificar para una presentación 510(k), pero se espera que la mayoría obtenga la aprobación previa a la comercialización.,
el proceso PMA requiere un estudio riguroso de un dispositivo médico para demostrar la seguridad y la eficacia a través del desarrollo de un perfil de beneficio/riesgo basado en datos. El proceso de PMA generalmente implica ensayos clínicos y un tiempo y recursos significativos para la recopilación de datos suficientes. Las únicas excepciones al proceso PMA dentro de la clase III son los dispositivos con un equivalente sustancial. Puede determinar si un dispositivo de clase III se puede comercializar con un 510 (k) buscando en la base de datos de aprobación previa a la comercialización (PMA) de la FDA y en la base de datos de notificación previa a la comercialización 510(k).,
Cómo determinar su clase
el primer paso para clasificar su dispositivo médico es navegar por las regulaciones de clasificación de la FDA, la lista de 16 categorías para Dispositivos Médicos de acuerdo con la especialización médica.
como ejemplo, le mostraremos los pasos para identificar la clasificación de una alarma de presión arterial. El dispositivo está clasificado en la categoría 870: dispositivos cardiovasculares.,
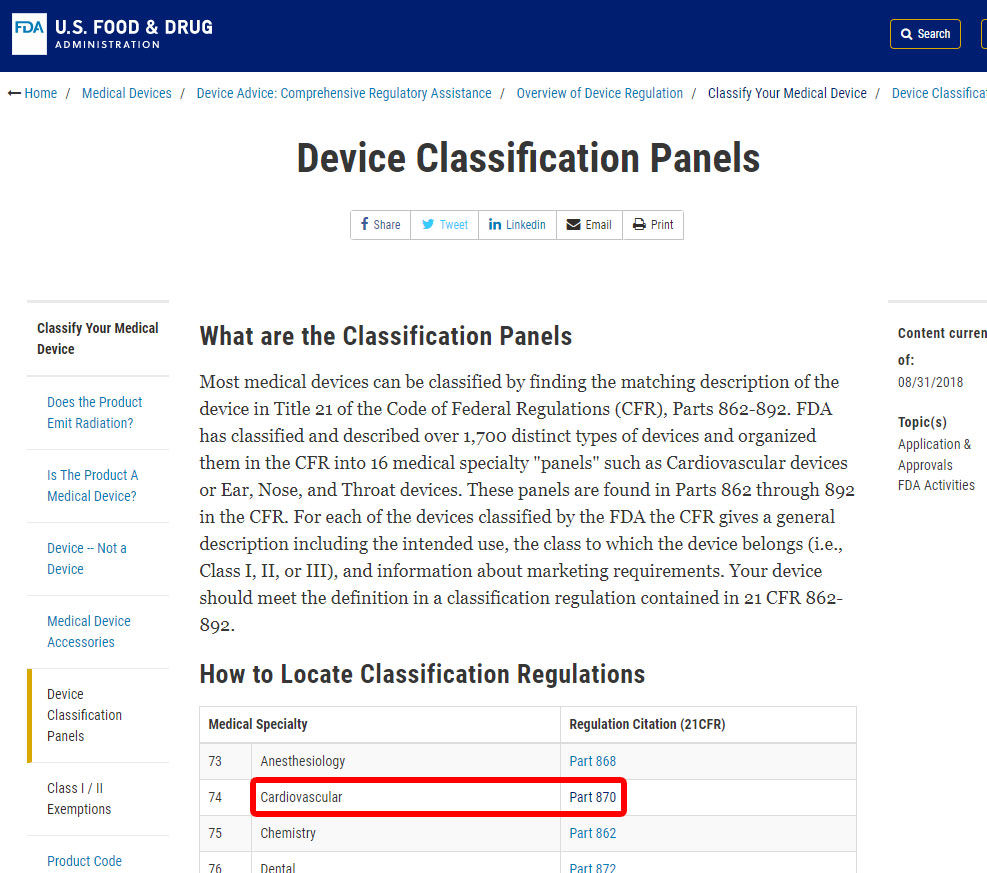
Una vez que haya localizado la especialidad médica relevante, haga clic en la categoría y navegue por la lista de dispositivos hasta encontrar un equivalente y el código de dispositivo asociado.
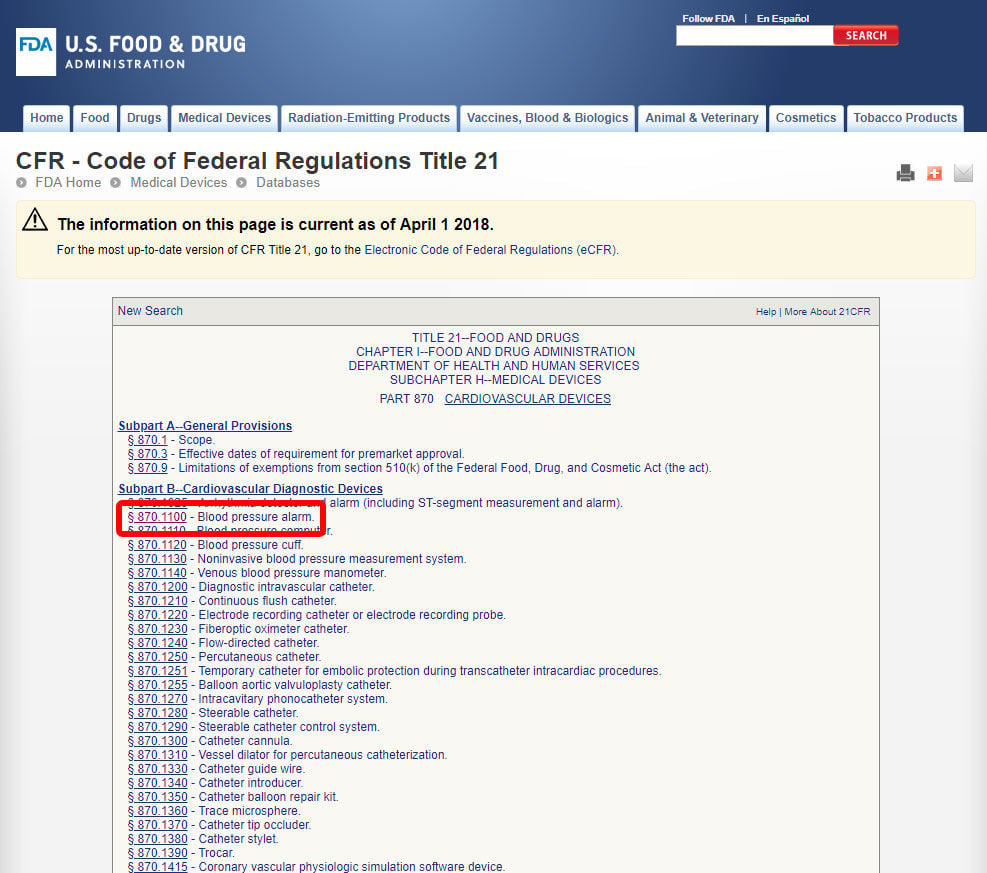
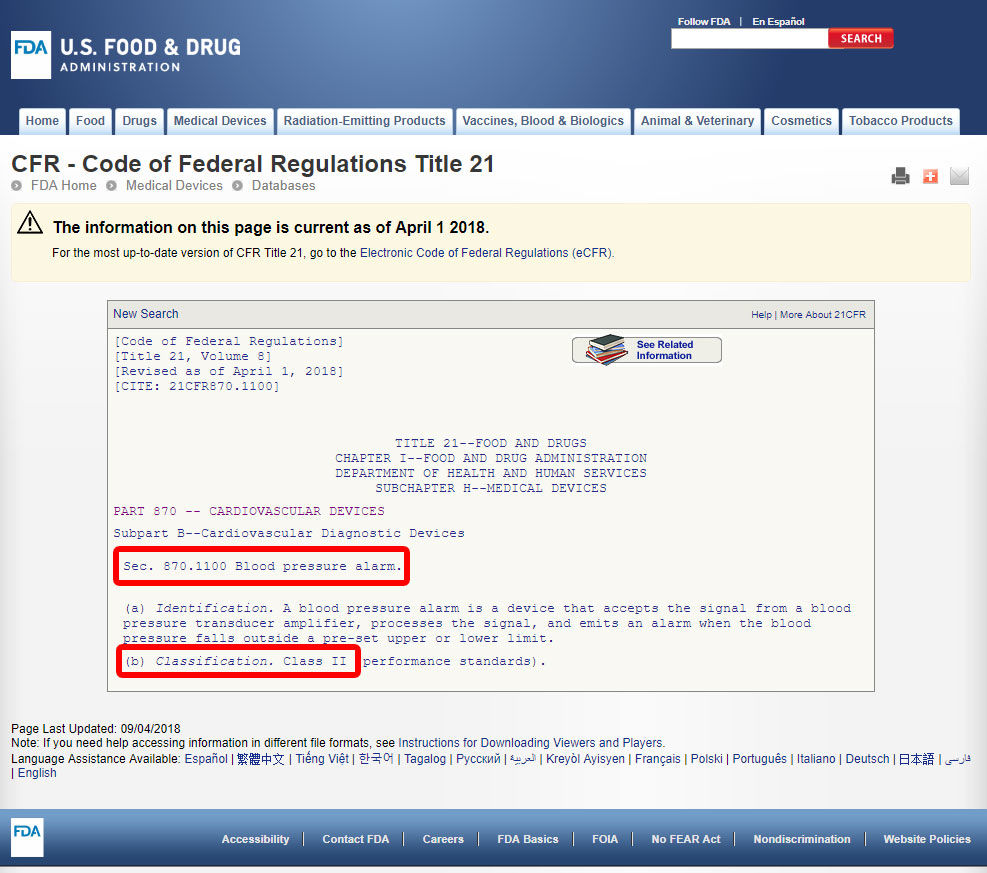
haga Clic en el código del dispositivo y abrir las directrices. La clasificación del dispositivo figura en la Sección (b).,
si su dispositivo carece de un equivalente listado entre los 1,700 dispositivos clasificados por la FDA, lo más probable es que sea un dispositivo innovador sin un equivalente sustancial y se clasificaría como clase III.
comprender las clases de Dispositivos Médicos de la FDA
Las diferencias entre los dispositivos médicos clasificados como clase I, II o III por la FDA y sus sistemas internos, y si un dispositivo es crítico para sostener la vida.,
además de estos factores, la FDA considera una equivalencia sustancial al determinar cómo se clasifica un dispositivo. Si su dispositivo es de bajo riesgo y se comunica mínimamente con el paciente, es probable que califique para la clase I y un proceso de aprobación del mercado simplificado. Los dispositivos de clase II y III deben demostrar la seguridad a través de la equivalencia sustantiva, una presentación 510(k) o el proceso de aprobación previa a la comercialización.
al saber cómo se clasifica su dispositivo, puede agilizar su camino hacia la aprobación del mercado al comprender los procesos y documentos que probablemente sean requeridos por la FDA., Si su organización está sujeta a un requisito de clase II o clase III 510(k) o PMA, este conocimiento puede ayudarlo a asignar los recursos apropiados por adelantado y planificar para una presentación exitosa.
