Holoprosencéphalie et strabisme
Pavlina S. Kemp, MD, Grant Casey, Susannah Q. Longmuir, MD
12 juin 2012
plainte principale: croisement des yeux
antécédents de maladie actuelle
La patiente est une femelle de 15 mois présentée à la clinique de l’œil, avec des antécédents d’hydrocéphalie sévère à la naissance. On lui a également diagnostiqué une holoprosencéphalie alobaire à la naissance avec des convulsions. Elle a été initialement référé pour eye crossing. Nous présentons son parcours clinique remarquable.,
antécédents médicaux:
- hydrocéphalie S/P shunt ventriculopéritonéal
- Holoprosencéphalie (type alobaire)
- Trouble convulsif
antécédents chirurgicaux:
- placement du shunt Ventriculopéritonéal, 2004
- révision du shunt Ventriculopéritonéal, 1/2005
- révision du shunt ventriculopéritonéal, 6/2005
antécédents familiaux: aucun antécédent familial connu D’Holoprosencéphalie, d’amblyopie ou de strabisme.
histoire sociale: le Patient vit à la maison avec ses parents et ses deux sœurs.,
médicaments: Aucun
examen et cours clinique:
âge: 15 mois
acuité visuelle: OD Centrale, instable et maintenue et OS central, instable et maintenu
Test D’acuité Teller:
- Sans correction OU: 20/800
élèves: également ronds et vivement réactifs, pas afférents relatifs défaut pupillaire.,
Vision stéréo: impossible de tester
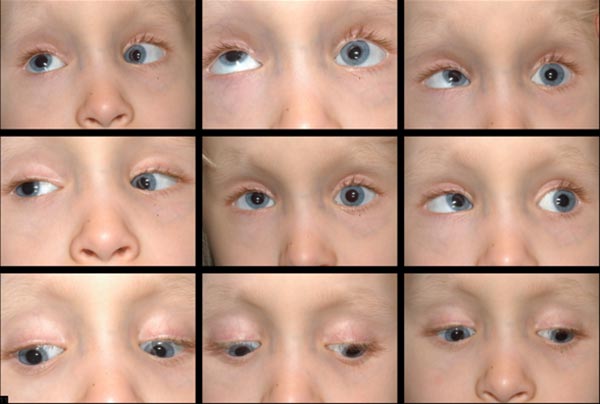
motilité et strabisme:
- grande ésotropie variable
- déficits bilatéraux d’élévation et d’abduction
- nystagmus horizontal Intermittent
réfraction Cycloplégique:
- OD: +4.00
- OS: +6.00
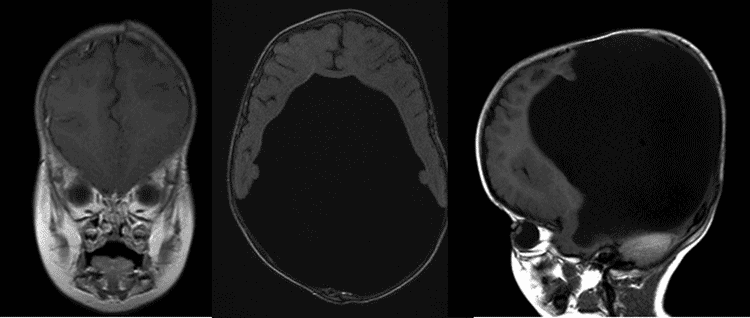
examen externe: remarquable pour une grande circonférence de la tête
examen de la lampe à fente: Examen normal du segment antérieur ou sans preuve de cataractes ou d’autres opacités médiatiques.
nerfs optiques apparents normaux et examen normal du fond d’œil dilaté. Aucun signe d’hypoplasie du nerf optique dans les deux yeux.,
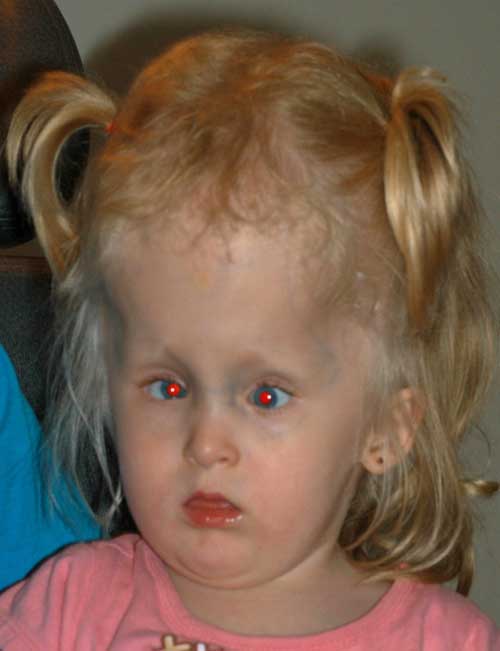
à ce stade, après discussion avec sa famille, la chirurgie du strabisme a été reportée et la correction de son hypermétropie a été tentée. Des lunettes ont été prescrites. Elle n’était pas en mesure de porter des lunettes confortablement et des lentilles de contact ont essayé. Des correctifs ont été effectués pour traiter son amblyopie. La patiente a été suivie et à l’âge de 3 ans, ses parents ont voulu procéder à une chirurgie du strabisme pour « décroiser” ses yeux.,
Age: 3
acuité visuelle: Central, CUSM OD et CUSM OS
motilité et strabisme:
- ésotropie grand angle de 55-60 dioptries prismatiques (Figure 2)
- déficits d’abduction bilatéraux et déficit d’élévation monoculaire gauche
Au moment de la chirurgie du strabisme, les ductions forcées peropératoires montraient des restrictions des deux muscles droits médiaux et le patient présentait une insertion anormale du muscle droit médial. L’insertion du muscle droit médial avant la désinsertion a été trouvée à 7 mm du limbe (plus postérieure que les 5,5 mm attendus du limbe). Secondaire à l’anatomie n’étant pas normale, une approche conservatrice a été effectuée et elle a subi des récessions médiales rectales bilatérales de 5,5 mm, laissant le droit médial à 12.,5 mm du limbe.
Après l’opération, elle présentait une petite ésotropie variable résiduelle avec une petite déviation verticale et une déficience en élévation plus perceptible de l’œil gauche.
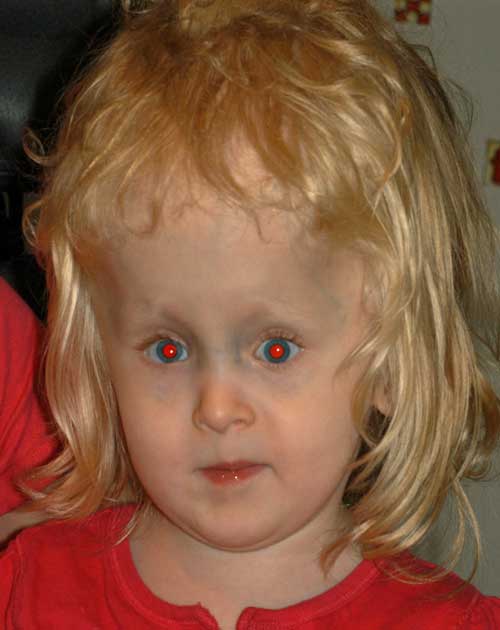
âge 6:
ses parents étaient satisfaits de l’alignement; cependant, avec le temps, elle a développé une hypertropie droite perceptible. À l’âge de 6 ans, sa famille a décidé de procéder à une deuxième chirurgie du strabisme pour remédier au désalignement vertical.,
une récession du droit inférieur gauche contre une récession du droit supérieur droit était prévue, la décision concernant la procédure à effectuer étant basée sur les ductions forcées peropératoires. Les ductions forcées Intra-opératoires ont montré un droit inférieur gauche serré, ce qui est compatible avec le déficit en élévation monoculaire que nous avions précédemment considéré. Une récession droite inférieure gauche de 6 mm a été réalisée, déplaçant le muscle de 8 mm postérieur au limbe, où il a été trouvé, à 14 mm postérieur au limbe.,
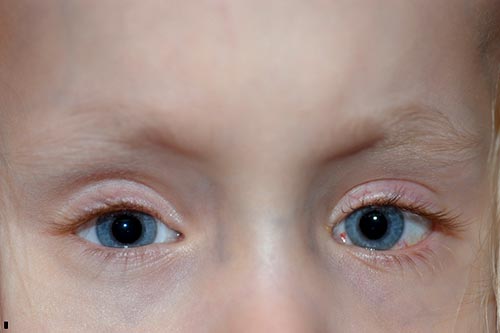
quelques mois après sa deuxième opération, sa mère a commencé à remarquer que son œil droit dérivait vers le haut en période d’inattention. Elle a été vue en clinique et a noté une déviation verticale dissociée manifeste à droite., Il a été décidé de procéder à nouveau avec une chirurgie du strabisme et une récession du droit supérieur droit de 8 mm par la méthode du repliement, déplaçant le muscle de 8 mm postérieur au limbe, où il a été trouvé, à 16 mm postérieur au limbe.
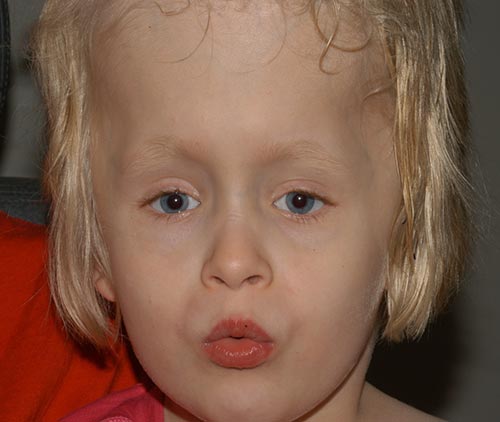
la patiente s’est bien comportée après l’opération (Figure 6) et a progressivement développé ses compétences visuelles. Elle a remarquablement bien fait dans l’ensemble et marche et lit maintenant.
diagnostic:
Holoprosencéphalie avec ésotropie infantile et strabisme complexe, avec variabilité des insertions du muscle droit médial.
Discussion:
L’Holoprosencéphalie est un type de trouble céphalique caractérisé par l’échec du prosencéphale (le cerveau antérieur embryonnaire) à se développer, conduisant à une structure cérébrale à un seul lobe et à de graves défauts crâniens et faciaux., Il existe trois classifications de l’holoprosencéphalie: l’holoprosencéphalie alobaire, l’holoprosencéphalie semi-lobaire et l’holoprosencéphalie lobaire.
l’holoprosencéphalie Alobaire représente les deux tiers des patients atteints, et est la forme la plus grave, caractérisée par l’échec du cerveau à se séparer en deux moitiés. Il en résulte un seul ventricule primitif, des bulbes olfactifs et des voies optiques absents et de graves anomalies du développement. Il est généralement associé à de graves anomalies faciales, notamment des yeux étroitement espacés, une petite taille de la tête, une fente labiale et palatine., L’holoprosencéphalie Semi-lobaire, qui représente un quart des cas d’holoprosencéphalie, est une forme intermédiaire de la maladie et se caractérise par des hémisphères cérébraux partiellement séparés et un seul ventricule. L’holoprosencéphalie lobaire est la forme la moins grave, dans laquelle le cerveau du patient peut être presque normal; il y a une fissure distincte entre les lobes centraux développés, et une certaine fusion des structures cérébrales est présente (Nanni, 2000). Dans la plupart des cas d’holoprosencéphalie, les malformations cérébrales sont incompatibles avec la vie., Dans les cas moins graves, les bébés naissent avec un développement cérébral normal ou presque normal et divers degrés de déformation faciale.
Les défauts craniofaciaux de la ligne médiane sont la caractéristique de l’holoprosencéphalie et peuvent inclure la microcéphalie, l’hypotélorisme (yeux anormalement rapprochés), les anomalies nasales, telles que l’aplatissement nasal ou un naris unique, et les défauts de la lèvre supérieure et du palais tels que la fente palatine ou une seule incisive avant. La cyclopie peut être présente dans les formes les plus sévères où une trompe en forme de nez est présente sur un seul œil au milieu du visage (Nanni, 2000)., On pense que le degré de déformation faciale indique la gravité des défauts intracrâniens. Les comorbidités associées comprennent un dysfonctionnement de l’hypophyse et de l’hypothalamus, entraînant une dérégulation de la température corporelle, des convulsions et un retard mental de gravité variable (Dubourg, 2007). Une hypotonie et une dystonie ont également été observées (Barkovich, 2002).
L’Holoprosencéphalie survient pendant les premières semaines de la vie intra-utérine. La prévalence de l’holoprosencéphalie au début du développement embryonnaire est de 1:250, diminuant à 1:10 000-1:20 000 à terme (Nanni, 2000)., Il n’y a pas de cause connue d’holoprosencéphalie, bien qu’il y ait eu de nombreux facteurs de risque suggérés, y compris le diabète maternel (risque de 1%, augmentation de 200 fois) (Barra, 1983), les infections pendant la grossesse, telles que les infections TORCH (Munke, 1989), et l’exposition à des substances toxiques, y compris l’alcool, le lithium, la Thorazine, les hormones, les anticonvulsivants et l’acide rétinoïque (Nanni, 2000). La plupart des cas sont considérés comme sporadiques, bien que l’holoprosencéphalie ait également une base génétique., On a observé une holoprosencéphalie familiale héritée à la fois autosomique dominante et autosomique récessive. Des anomalies chromosomiques ont également été associées à l’holoprosencéphalie, la trisomie 13 étant la plus fréquente, bien qu’il ne s’agisse pas d’une association constante (Kallen, 1992).
on pense que la pathogenèse implique un défaut dans les gènes de signalisation responsables de la régulation de la structure du tube neural., Les résultats intracrâniens comprennent une hypoplasie corticale variable, une fusion variable du diencéphale, des ganglions de la base et du thalamus et la présence d’un kyste dorsal (résultant d’une thalami fusionnée) en expansion à partir d’un 3e ventricule partiellement bloqué (Simon, 2001). L’hydrocéphalie, causée par une accumulation anormale de LCR dans les ventricules, n’est pas rare dans l’holoprosencéphalie et serait due à une malformation des ventricules ou à une production excessive de LCR., Cela complique souvent la classification de l’holoprosencéphalie, car le cerveau est comprimé et le crâne précédemment microcéphale est autorisé à se dilater avant la fusion des sutures crâniennes (Tripathi, 2009). Il est important d’aborder la vision du patient pour permettre une interaction optimale avec son environnement. Souvent, les lunettes ne sont pas tolérées en raison du degré d’asymétrie faciale et d’anomalies structurelles présentes. Dans ces situations, nous pensons que les lentilles de contact doivent être considérées comme un moyen d’améliorer la fonction visuelle.,
bien qu’il existe une variation de l’insertion du muscle droit médial dans la population générale, ce cas est remarquable pour les insertions anormales du muscle extraoculaire, en particulier la distance de 7 mm du recti médial du limbe. Comme mentionné ci-dessus, les défauts de la ligne médiane sont fréquents dans l’holoprosencéphalie, ce qui peut expliquer pourquoi les recti médiaux étaient préférentiellement impliqués. À l’exception de la cyclopie, peu de publications ont été publiées sur les associations oculaires et strabismiques avec l’holoprosencéphalie.,
dans l’ensemble, le traitement est hautement individualisé en fonction de la gravité du patient et de la configuration de la malformation. Le traitement est favorable et symptomatique et le pronostic dépend grandement du type d’holoprosencéphalie et des anomalies associées (Nanni, 2000).
dans le cas présenté ci-dessus, l’une des considérations les plus importantes pour la famille du patient était de savoir comment intervenir pour améliorer la vision et permettre une croissance et un développement continus. Le patient était gravement malade au cours des premières années de sa vie, et les soins palliatifs ont d’abord été discutés comme une option viable., La famille du patient voulait poursuivre le traitement et rechercher des interventions possibles pour améliorer la qualité de vie. Face à des situations difficiles comme celle-ci, il est essentiel que les fournisseurs de soins de santé aident les familles à prendre des décisions dans un environnement respectueux, sans jugement ni influence indue. Le groupe de travail de L’American College of Critical Care Medicine a publié des lignes directrices de pratique clinique traitant du soutien de la famille dans l’Unité de soins intensifs centrée sur le patient (Davidson, 2007)., Grâce à une bonne communication, à la gestion des conflits et à la facilitation des réunions, les familles peuvent être impliquées dans un modèle de prise de décision partagée dans lequel les familles ne sont pas les seules responsables de toutes les décisions médicales de manière autonome, pas plus que les fournisseurs de soins médicaux ne fournissent des soins paternalistes. Lors des réunions familiales, il est recommandé de poser aux membres de la famille des questions ouvertes sur leur compréhension des soins du patient, leurs peurs et leurs stratégies d’adaptation., Les fournisseurs de soins sont ensuite encouragés à répéter les sentiments de la famille pour permettre le développement de la confiance dans l’équipe et le processus décisionnel. Après cela, les praticiens doivent fournir des informations claires et honnêtes dans un langage accessible, avec la possibilité de poser des questions. Le but de la discussion est le consensus, qui est aidé par la reconnaissance respectueuse de toutes les opinions.,
Penticuff et Arheart ont étudié l’efficacité des rencontres entre les fournisseurs de soins de santé et les parents dans une unité de soins intensifs néonatals et ont montré que la prise de décision partagée entraînait moins de conflits, des attentes parentales irréalistes et une meilleure collaboration, tout en aidant les parents à mieux comprendre la situation médicale de leur enfant (Penticuff, 2005). Les niveaux de stress de la famille sont démontrés être diminués avec une communication ouverte et efficace, ainsi qu’un environnement d’espoir (Davidson, 2007)., Il est important de noter que de tels soins centrés sur le patient améliorent également les résultats cliniques (Lewin, 2001).
Diagnostic Différentiel
- Esotropia
- Monoculaire élévation carence
- déviation verticale Dissociée
Résumé
|
signes
|
symptômes
|
traitement
|
barkovich AJ, Simon em, Clegg NJ, kinsman SL, Hahn js. Analyse du cortex cérébral dans l’holoprosencéphalie avec attention aux fissures sylviennes. AJNR Am J Neuroradiol. 2002;23(1):143-50.
Blaas HG, Eriksson AG, Salvesen ka, Isaksen CV, Christensen B, Møllerløkken G, Eik-Nes SH., Cerveaux et visages dans l’holoprosencéphalie: description pré et postnatale de 30 cas. Échographie Obstet Gynecol. 2002;19(1):24-38.
Barra Jr M, Hanson JW, Currey K, Sharp S, Toriello H, Schmickel RD, Wilson GA. Holoprosencéphalie chez les nourrissons de mères diabétiques. J Pediatr 1983; 102: 565D8.
Davidson JE, Powers K, Hedayat KM, Tieszen M, Kon AA, Shepard E, Spuhler V, Todres ID, Levy M, Barr J, Ghandi R, Hirsch G, Armstrong D., Lignes directrices de pratique clinique pour le soutien de la famille dans l’approche centrée sur le patient en unité de soins intensifs: Collège Américain de Médecine de Soins intensifs Task Force 2004-2005. Collège américain de Médecine de Soins intensifs Task Force 2004-2005, la Société de Médecine de Soins Critiques. Crit Care Med. 2007;35(2):605-22.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent s, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007 2;2:8.
Kallen B, Castilla EE, Lancaster PAL, et coll. Le cyclope et la sirène: une étude épidémiologique de deux types de malformation rare. J Med Genet 1992; 29:30-35.,
Lewin SA, Skea ZC, Entwistle V, Zwarenstein M, Dick J. Interventions pour les fournisseurs afin de promouvoir une approche centrée sur le patient dans les consultations cliniques. Cochrane Database Syst Rev. 2001; (4): CD003267. (PMID:11687181)
Munke M. Clinique, cytogénétique et moléculaire des approches de l’hétérogénéité génétique de l’holoprosencéphalie. Am J Med Genet 1989; 34: 237-245.
Nanni L, Schelper RL, Muenke MT. Génétique moléculaire de l’holoprosencéphalie. Avant Biosci. 2000 1; 5: D334-42.
Penticuff JH, Arheart KL.,Efficacité d’une intervention pour améliorer la collaboration parents-professionnels en soins intensifs néonatals. J Perinat Nurs Néonatales. 2005;19(2):187-202.
Scott WE, Jackson OB. Paralysie à double ascenseur: l’importance de la restriction du droit inférieur. Suis Orthopt J. 1977;27:5-10.
Simon EM, HEVNER RF, Pinter J, Clegg NJ, Delgado M, Kinsman SL, Hahn JS, Barkovich AJ. La dorsale kyste dans holoprosencéphalie et le rôle du thalamus dans sa formation. La neuroradiologie. 2001;43(9):787-91.
Tripathi AK, Agrawal D, Sedain G. hydrocephalic holoprosencephaly: an oxymoron?, Aperçu de l’étiologie et de la gestion. J Pediatr Neurosci. 2009;4(1):41-3.
consentement pour l’utilisation de photos et de vidéos obtenu de la mère du patient.
format de Citation suggéré: Kemp PS, Casey, G, LONGMUIR SQ. Holoprosencéphalie et Strabismus.Eyerounds.org. 12 juin 2012, disponible auprès de: http://EyeRounds.org/cases/151-holoprosencephaly-strabismus.htm