Quelles sont les différences dans les classes de dispositifs médicaux de la FDA?
La FDA réglemente tous les dispositifs médicaux commercialisés aux états-unis, qui sont regroupés en trois grandes catégories. Tout dispositif médical approuvé PAR LA FDA est classé dans la classe I, II ou III en fonction du risque du dispositif, de son caractère invasif et de son impact sur la santé globale du patient. Mais où sont les lignes tracées entre chacune de ces trois classes, et pourquoi?,
les directives de classification de la FDA des États-Unis peuvent être très déroutantes pour les fabricants de dispositifs médicaux ayant une exposition limitée au système. Il y a une énorme différence dans le chemin optimal vers le marché pour les fabricants en fonction de la façon dont votre appareil est groupé. Les dispositifs de classe I sont soumis à beaucoup moins d’exigences réglementaires que les dispositifs de classe II ou III.
en comprenant les différences dans les classes de dispositifs médicaux de la FDA, vous pouvez comprendre comment votre dispositif sera regroupé., Grâce à ces connaissances, les fabricants de dispositifs médicaux en phase de pré-commercialisation peuvent mieux préparer et allouer les ressources nécessaires à l’approbation réglementaire.
différences entre les Classes de dispositifs médicaux de la FDA
la FDA a classé plus de 1 700 types distincts de dispositifs médicaux. Les appareils sont organisés dans le Code of Federal Regulations (CFR) selon 16 spécialités, telles que les appareils cardiovasculaires ou hématologiques., La classification de votre dispositif médical selon l’une des 16 spécialités est la première étape pour savoir si vous fabriquez un dispositif médical de classe I, II ou III.
Après avoir classé un appareil en fonction de sa spécialité, la FDA demande aux fabricants de procéder à la notification avant la mise sur le marché en sachant si leur appareil est exempté ou non. Les dispositifs médicaux de classe I, la catégorie la moins risquée et la moins invasive, sont exemptés des processus de notification avant la mise en marché. Les dispositifs spécifiques de classe II sont également exemptés de l’approbation avant la mise en marché.,
cependant, tous les appareils réglementés par la FDA sont soumis aux exigences actuelles de bonnes pratiques de fabrication (cGMP) en matière d’enregistrement, d’étiquetage et de qualité. Mais comment savoir si votre appareil est de classe I ou II et si vous devez subir une notification avant la mise sur le marché? 
Classe 1
la FDA des États-Unis définit les dispositifs de classe I comme des dispositifs qui » ne sont pas destinés à être utilisés pour soutenir ou maintenir la vie ou d’une importance substantielle dans la prévention des atteintes à la santé humaine, et ils peuvent ne pas, »
ces appareils sont la classe la plus courante d’appareils réglementés par la FDA, constituant 47% des appareils approuvés sur le marché.
Les dispositifs de classe I ont un contact minimal avec les patients et un faible impact sur la santé globale du patient. En général, les dispositifs de classe I n »entrent pas en contact avec les organes internes d » un patient, le système nerveux central, ou le système cardiovasculaire. Ces appareils sont soumis au moins d’exigences réglementaires.,
exemples de dispositifs de classe I:
- Brosse À Dents Électrique
- abaisse-langue
- Masque À Oxygène
- Scalpel chirurgical réutilisable
- Bandages
- lits D’Hôpital
mise sur le marché de dispositifs médicaux de classe I
les dispositifs de classe I sont les plus rapides et les plus faciles à essentiel aux soins de maintien de la vie. La majorité des dispositifs de classe I sont exemptés des exigences de la FDA en matière de Notification avant la mise sur le marché (510K) et D’approbation avant la mise sur le marché (PMA).,
Les dispositifs de classe I ne sont pas exemptés des contrôles généraux de la FDA, une série de commandes qui s’applique aux dispositifs médicaux de classe I, II et III. Les dispositions de cette loi traitent de l’adultération, de la mauvaise marque, de l’enregistrement des appareils, des dossiers et des bonnes pratiques de fabrication. Les fabricants de dispositifs médicaux qui entrent dans la Classe A sont toujours tenus de mettre en œuvre un système de gestion de la qualité et de suivre les normes pour garantir un produit de qualité.,
lecture connexe: la différence entre la Notification avant mise en marché 510(k) et L’approbation avant mise en marché
Classe 2
Les instruments médicaux de classe II sont plus compliqués que les instruments de classe I et présentent une catégorie de risque plus élevée parce qu’ils sont plus susceptibles d’entrer en contact Cela peut inclure des dispositifs qui entrent en contact avec le système cardiovasculaire ou les organes internes d »un patient, et des outils de diagnostic.,
la FDA définit les dispositifs de classe II comme des « dispositifs pour lesquels les contrôles généraux sont insuffisants pour fournir une assurance raisonnable de leur sécurité et de leur efficacité.,
mise sur le marché de dispositifs médicaux de classe II
Les contrôles varient en fonction du dispositif, mais selon la FDA, peuvent inclure:
- performance de L’appareil
- surveillance après la mise/li>
- exigences spéciales en matière D’étiquetage
- exigences relatives aux données avant la mise sur le marché
- lignes directrices
la majorité des dispositifs de classe II sont approuvés par la FDA pour le marché par le biais du processus de notification avant la mise sur le marché, ou 510(K).,
Les dispositifs de classe II sont soumis aux mêmes contrôles généraux mentionnés ci-dessus, mais la FDA Les définit comme étant des « dispositifs pour lesquels les contrôles généraux sont insuffisants pour fournir une assurance raisonnable de la sécurité et de l’efficacité du dispositif.” Pour cette raison, les appareils de classe II sont également soumis à des contrôles spéciaux. Ces réglementations dépendent de l’appareil et peuvent inclure des exigences d’étiquetage spéciales, des registres de patients et des normes de performance.
la plupart des appareils de classe II sont commercialisés en utilisant le processus de Notification avant commercialisation (510K)., Le 510 (k) est une application complexe à la FDA, qui démontre qu’un dispositif est sûr et efficace en démontrant que le dispositif est équivalent à un autre dispositif qui est sur le marché.
Ce processus consiste à montrer « l’équivalence substantielle » à un autre dispositif qui est connu dans le langage FDA comme « le prédicat. »Cela ne signifie pas que les appareils doivent être identiques, mais ils nécessitent des similitudes significatives dans l » utilisation, conception, matériaux, étiquetage, Normes, et d » autres caractéristiques.,
la FDA a publié une liste d’exemption au début de 2018 qui exempte plus de 800 instruments médicaux génériques de classe I et II du processus 510(k). Si vous avez un dispositif médical générique de classe II, vous pouvez savoir s’il est exempté d’un dépôt 510(k) en effectuant une recherche dans la base de données de Classification des produits de la FDA.
lecture connexe: 5 Raisons la révision de FDA 510(k) est une excellente décision.
Classe 3
la FDA définit les dispositifs de classe III comme des produits qui « supportent généralement la vie, sont implantés ou présentent un risque potentiel déraisonnable de maladie ou de blessure., »
seulement 10% des appareils réglementés par la FDA américaine entrent dans la classe III. cette classification est généralement étendue aux implants permanents, aux dispositifs médicaux intelligents et aux systèmes de survie.
bien que la classe III soit généralement réservée aux dispositifs médicaux les plus innovants et de pointe, il existe d’autres dispositifs qui peuvent entrer dans la classe III pour différentes raisons., Certains instruments classés initialement dans la classe II peuvent être classés dans la classe III si le fabricant n’est pas en mesure de démontrer une équivalence substantielle à un prédicat (produit existant) au cours du processus de dépôt de la PMA (510K).,
exemples de dispositifs médicaux de classe III:
- implants mammaires
- stimulateurs cardiaques
- défibrillateurs
- ventilateurs à haute fréquence
- implants cochléaires
- moniteurs de prélèvement de sang fœtal
- prothèses implantées
mise sur le marché de dispositifs médicaux de classe III
Les dispositifs de classe III sont les contrôles et le processus d’approbation avant commercialisation (PMA) de la FDA., La FDA écrit: « en raison du niveau de risque associé aux dispositifs de classe III, la FDA a déterminé que les contrôles généraux et spéciaux seuls sont insuffisants pour assurer la sécurité et l’efficacité des dispositifs de classe III. »
la PMA est le type le plus intensif d’application de commercialisation d’appareils requis par la FDA. Certains dispositifs de classe III de la FDA sont exemptés et peuvent être admissibles à un dépôt 510(k), mais la majorité devrait obtenir l’approbation avant la mise en marché.,
le processus de PMA nécessite une étude rigoureuse d’un dispositif médical pour prouver l’innocuité et l’efficacité grâce à l’élaboration d’un profil avantages/risques fondé sur les données. Le processus de PMA implique généralement des essais cliniques et beaucoup de temps et de ressources pour une collecte de données suffisante. Les seules exceptions au processus PMA dans la classe III sont les dispositifs avec un équivalent substantiel. Vous pouvez déterminer si un appareil de classe III Peut être commercialisé avec un 510 (k) en effectuant une recherche dans la base de données D’approbation préalable à la mise en marché (PMA) de la FDA et dans la base de données de Notification préalable à la mise en marché 510(k).,
Comment déterminer votre classe
la première étape vers la classification de votre dispositif médical est de naviguer dans les règlements de classification de la FDA, la liste des 16 catégories pour les dispositifs médicaux selon la spécialisation médicale.
Comme exemple, nous allons vous montrer les étapes pour identifier la classification de la pression artérielle d’alarme. L’appareil est classé dans la catégorie 870: appareils Cardiovasculaires.,
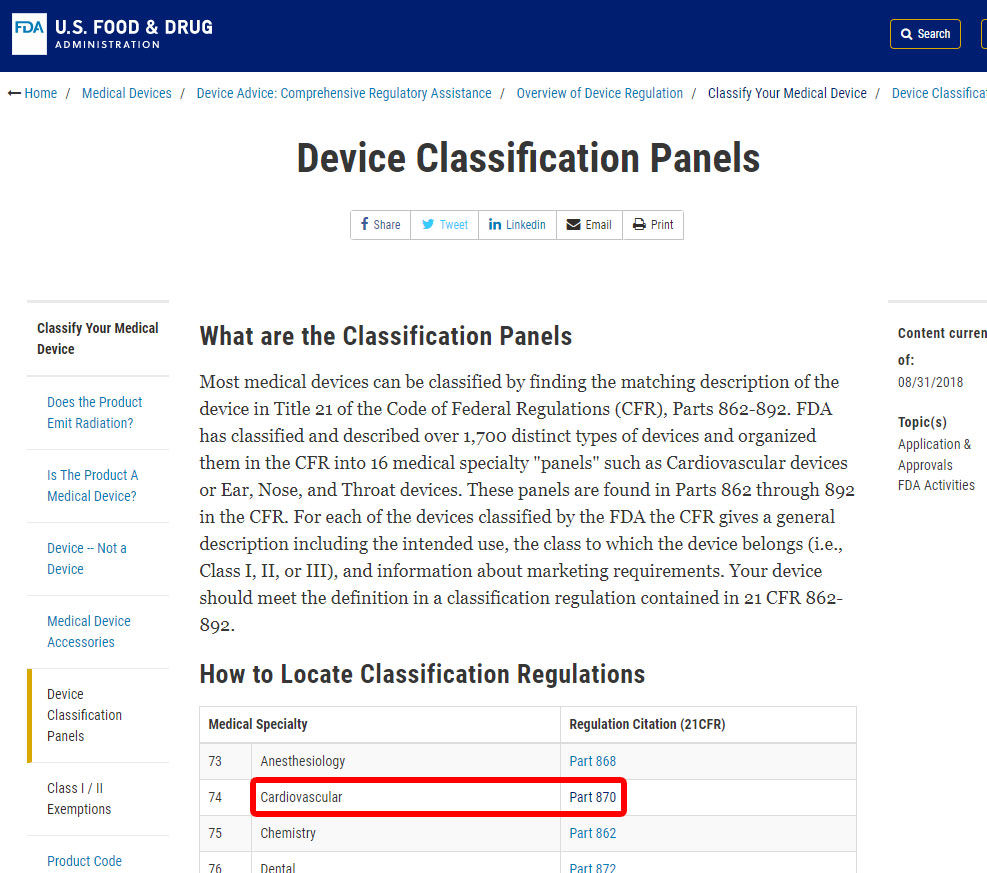
Une fois que vous avez trouvé la spécialité médicale pertinente, cliquez sur la catégorie et naviguez dans la liste des appareils jusqu’à ce que vous trouviez un équivalent et le code de l’appareil associé.
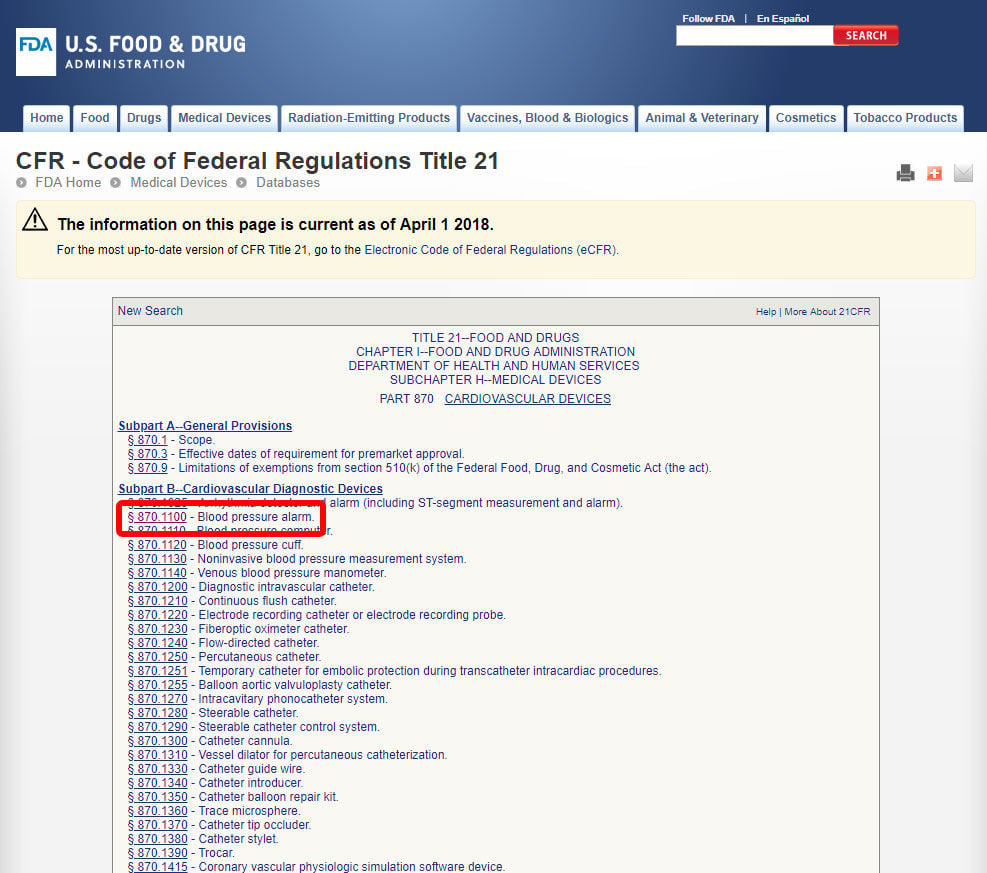
Cliquez sur le code de l’appareil et ouvrir les lignes directrices. La classification de l’appareil est indiquée à la section b).,
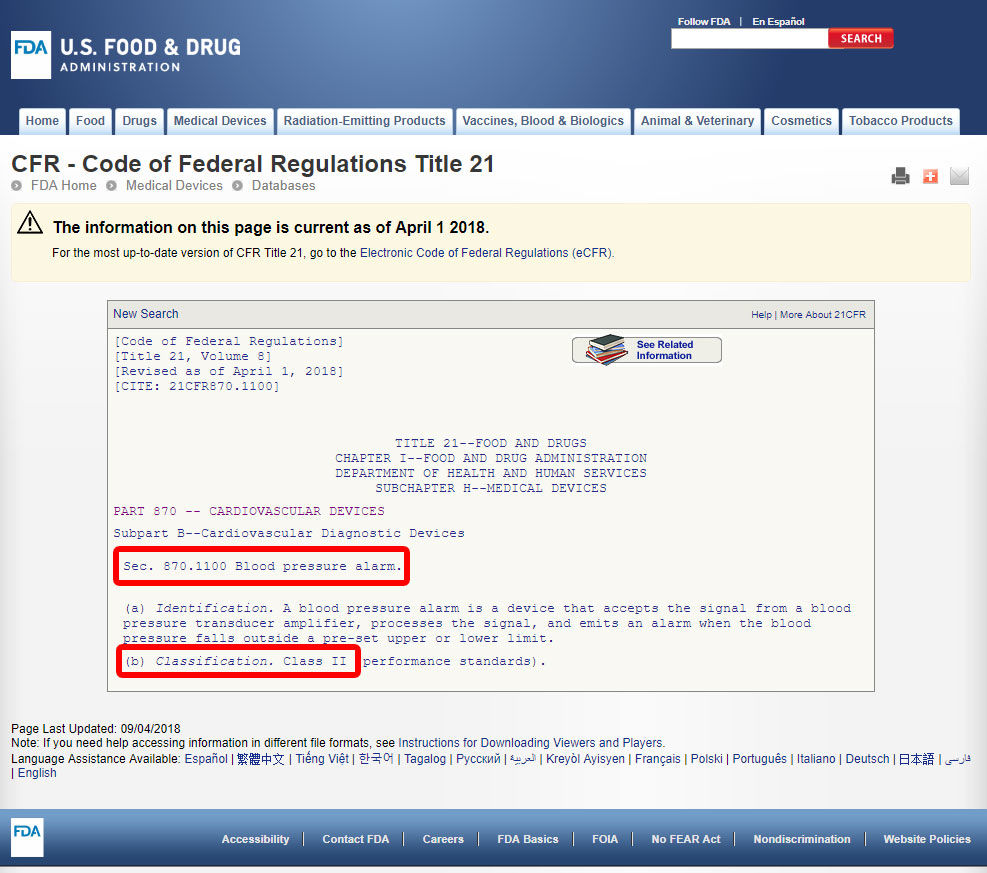
Si votre appareil n’a pas d’équivalent répertorié parmi les 1 700 appareils Classés par la FDA, il s’agit très probablement d’un appareil innovant sans équivalent substantiel et serait classé dans la classe III.
comprendre les Classes de dispositifs médicaux de la FDA
Les différences entre leurs systèmes internes, et si un appareil est essentiel au maintien de la vie.,
en plus de ces facteurs, la FDA tient compte de l’équivalence substantielle lors de la détermination de la classification d’un instrument. Si votre appareil présente un faible risque et communique avec le patient de manière minimale, vous serez probablement admissible à la classe I et à un processus simplifié d’approbation du marché. Les dispositifs de classe II et III doivent démontrer leur sécurité au moyen d’une équivalence de fond, d’un dépôt 510(k) ou du processus d’approbation avant la mise en marché.
en sachant comment votre appareil est classé, vous pouvez rationaliser votre chemin vers l’approbation du marché en comprenant les processus et les documents qui sont susceptibles d’être requis par la FDA., Si votre organisation est assujettie à une exigence de classe II ou de classe III 510(k) ou de PMA, ces connaissances peuvent vous aider à allouer les ressources appropriées à l’avance et à planifier un dépôt réussi.
