Oloprosencefalia e strabismo
Pavlina S. Kemp, MD, Grant Casey, Susannah Q. Longmuir, MD
12 giugno 2012
Denuncia principale: Eye crossing
Storia della malattia attuale
Il paziente è una femmina di 15 mesi alla presentazione alla clinica oculistica, con una storia di idrocefalo grave alla nascita. Le è stata anche diagnosticata alobar oloprosencefalia alla nascita con convulsioni. Lei è stato originariamente indicato per crossing occhio. Vi presentiamo il suo notevole corso clinico.,
Storia Medica:
- Idrocefalo s/p ventriculoperitoneal manovra
- Oloprosencefalia (alobar tipo)
- epilessia
la Storia Passata Chirurgica:
- shunt ventricolo-peritoneale di collocamento, 2004
- shunt ventricolo-peritoneale di revisione, 1/2005
- shunt ventricolo-peritoneale di revisione, 6/2005
Storia di Famiglia: Non sono noti storia familiare di oloprosencefalia, ambliopia o strabismo.
Storia sociale: Il paziente vive a casa con i genitori e due sorelle.,
Farmaci: Nessuno
Esame e decorso clinico:
Età: 15 mesi
Acuità visiva: Centrale, instabile e mantenuta OD e centrale, instabile e mantenuta OS
Test di acuità del cassiere:
- Senza correzione OU: 20/800
Alunni: ugualmente rotondi e vivacemente reattivi, senza afferenti difetto pupillare.,
la Stereo Visione: Impossibile per il test
la Motilità e Strabismo:
- variabile di Grandi dimensioni esotropia
- Bilaterali elevazione e il rapimento deficit
- Intermittente nistagmo orizzontale
Cycloplegic Rifrazione:
- OD: +4.00
- OS: +6.00
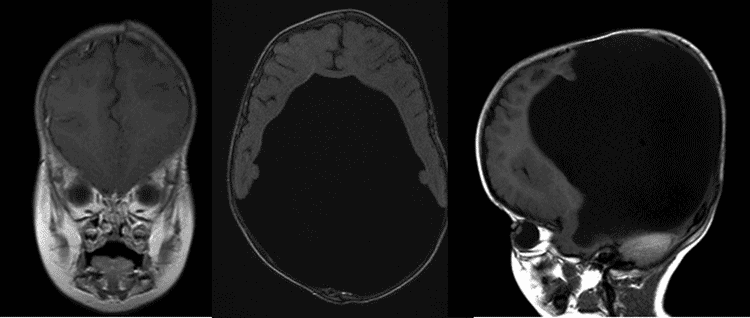
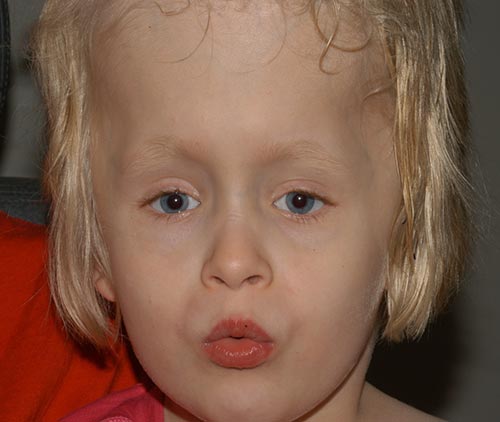
Esterni Esame: Notevole per la grande circonferenza della testa
Lampada a Fessura Esame: Normale segmento anteriore dell’esame OU senza evidenza di cataratta o di altri supporti di opacità.
Normali nervi ottici apparenti e normale esame del fondo dilatato. Nessun segno di ipoplasia del nervo ottico in entrambi gli occhi.,
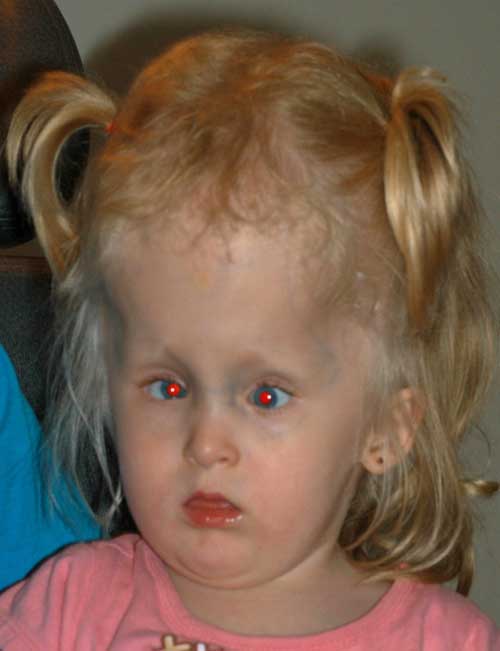
A questo punto, dopo una discussione con la sua famiglia, l’intervento chirurgico per lo strabismo è stato rinviato e la correzione della sua ipermetropia è stata tentata. Gli occhiali sono stati prescritti. Non era in grado di indossare gli occhiali comodamente e le lenti a contatto sono state provate. La patch è stata eseguita per trattare la sua ambliopia. La paziente è stata seguita e all’età di 3 anni, i suoi genitori volevano procedere con la chirurgia dello strabismo per “uncross” i suoi occhi.,
Età: 3
l’Acuità Visiva Centrale, CUSM OD e CUSM OS
la Motilità e Strabismo:
- Grande angolo di esotropia di 55-60 prisma diottrie (Figura 2)
- Bilaterali rapimento deficit e a sinistra monoculare elevazione carenza
Al momento della chirurgia dello strabismo, le duzioni forzate intraoperatorie mostravano restrizioni di entrambi i muscoli retti mediali e il paziente risultava avere un’inserzione anomala del muscolo retto mediale. L’inserzione del muscolo retto mediale prima della disinserzione è stata trovata a 7 mm dal limbus (più posteriore del previsto 5,5 mm dal limbus). Secondaria all’anatomia non normale, è stato eseguito un approccio conservativo e ha subito recessioni del retto mediale bilaterale di 5,5 mm, lasciando il retto mediale a 12.,5 mm dal limbus.
Post-operatoria, aveva una piccola esotropia variabile residua con una piccola deviazione verticale e una carenza di elevazione più evidente dell’occhio sinistro.
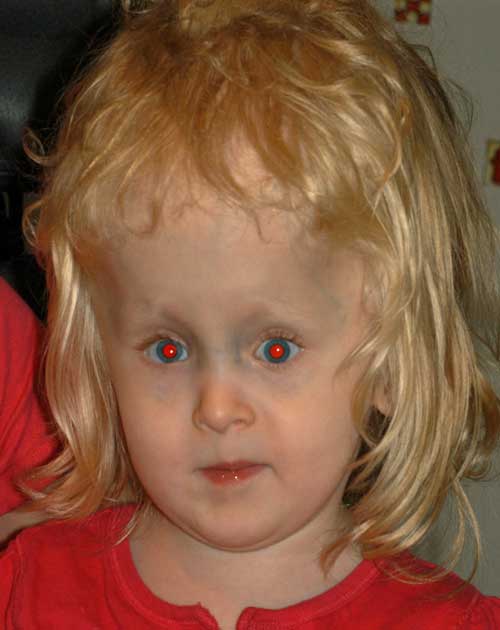
Età 6:
I suoi genitori erano felici con l’allineamento; tuttavia, con il tempo, ha sviluppato una notevole ipertrofia destra. All’età di 6 anni, la sua famiglia ha deciso di procedere con un secondo intervento di strabismo per affrontare il disallineamento verticale.,
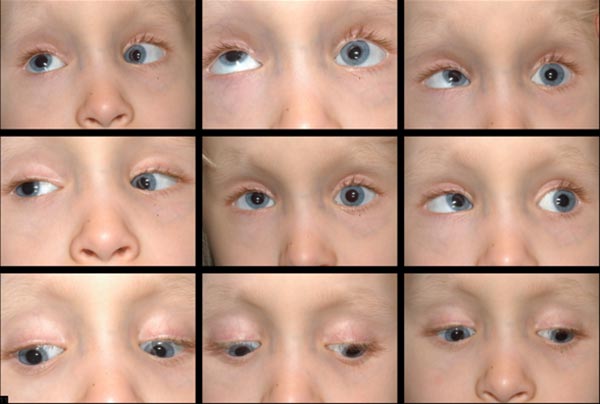
È stata pianificata una recessione del retto inferiore sinistro rispetto a una recessione del retto superiore destro, con decisione su quale procedura sarebbe stata eseguita in base alle condotte forzate intraoperatorie. Le canalizzazioni forzate intraoperatorie hanno mostrato un retto inferiore sinistro stretto, che è coerente con la carenza di elevazione monoculare che avevamo precedentemente considerato. È stata eseguita una recessione del retto inferiore sinistro di 6 mm, spostando il muscolo da 8 mm posteriore al limbus, dove è stato trovato, a 14 mm posteriore al limbus.,
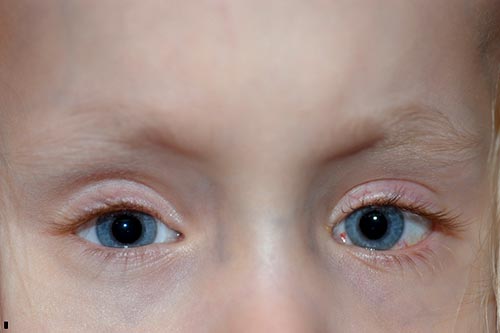
Pochi mesi dopo il suo secondo intervento, sua madre cominciò a notare che il suo occhio destro si spostava verso l’alto in momenti di disattenzione. È stata vista in clinica e ha notato una deviazione verticale dissociata manifesta sulla destra., Si è deciso di procedere nuovamente con la chirurgia dello strabismo e una recessione del retto superiore destro di 8 mm con il metodo hangback, spostando il muscolo da 8 mm posteriore al limbus, dove è stato trovato, a 16 mm posteriore al limbus.
La paziente ha fatto bene post-operatorio (Figura 6), e sta progressivamente sviluppando le sue capacità visive. Ha fatto molto bene nel complesso e sta camminando e ora leggendo.
Diagnosi:
Oloprosencefalia con esotropia infantile e strabismo complesso, con variabilità nelle inserzioni muscolari del retto mediale.
Discussione:
L’oloprosencefalia è un tipo di disturbo cefalico caratterizzato dal fallimento del prosencefalo (il proencefalo embrionale) a svilupparsi, portando a una struttura cerebrale a lobo singolo e gravi difetti del cranio e del viso., Ci sono tre classificazioni di oloprosencefalia: oloprosencefalia alobare, oloprosencefalia semi-lobare e oloprosencefalia lobare.
L’oloprosencefalia di Alobar rappresenta due terzi dei pazienti affetti ed è la forma più grave, caratterizzata dall’incapacità del cervello di separarsi in due metà. Ciò si traduce in un singolo ventricolo primitivo, bulbi olfattivi assenti e tratti ottici e gravi anomalie dello sviluppo. Di solito è associato a gravi anomalie facciali, tra cui occhi ravvicinati, piccole dimensioni della testa, labbro leporino e palato., L’oloprosencefalia semi-lobare, che rappresenta un quarto dei casi di oloprosencefalia, è una forma intermedia della malattia ed è caratterizzata da emisferi cerebrali parzialmente separati e da un singolo ventricolo. L’oloprosencefalia lobare è la forma meno grave, in cui il cervello del paziente può essere quasi normale; c’è una fessura distinta tra i lobi centrali sviluppati e una certa fusione delle strutture cerebrali è presente (Nanni, 2000). Nella maggior parte dei casi di oloprosencefalia, le malformazioni cerebrali sono incompatibili con la vita., Nei casi meno gravi, i bambini nascono con uno sviluppo cerebrale normale o quasi normale e vari gradi di deformità facciale.
I difetti craniofacciali della linea mediana sono il segno distintivo dell’oloprosencefalia e possono includere microcefalia, ipotelorismo (occhi anormalmente ravvicinati), anomalie nasali, come appiattimento nasale o un singolo naris, e difetti del labbro superiore e del palato come palatoschisi o un singolo incisivo anteriore. La ciclopia può essere presente nelle forme più gravi in cui una proboscide simile al naso è presente su un singolo occhio al centro del viso (Nanni, 2000)., Il grado di deformità facciale è pensato per indicare la gravità dei difetti intracranici. Le comorbilità associate includono la disfunzione della ghiandola pituitaria e dell’ipotalamo, con conseguente disregolazione della temperatura corporea, convulsioni e ritardo mentale di varia gravità (Dubourg, 2007). Sono state osservate anche ipotonia e distonia (Barkovich, 2002).
L’oloprosencefalia si verifica durante le prime settimane di vita intrauterina. La prevalenza di oloprosencefalia nello sviluppo embrionale precoce è 1: 250, diminuendo a 1:10.000-1:20.000 a termine (Nanni, 2000)., Non esiste una causa nota di oloprosencefalia, anche se ci sono stati molti fattori di rischio suggeriti, tra cui il diabete materno (rischio 1%, aumento di 200 volte) (Barra, 1983), infezioni durante la gravidanza, come le infezioni TORCH (Munke, 1989) e l’esposizione a sostanze tossiche, tra cui alcol, litio, torazina, ormoni, anticonvulsivanti e acido retinoico (Nanni, 2000). La maggior parte dei casi sono considerati sporadicamente, anche se oloprosencefalia è stato trovato per avere una base genetica pure., L’oloprosencefalia familiare è stata osservata ereditata sia nei modelli autosomici dominanti che autosomici recessivi. Le anomalie cromosomiche sono state anche associate all’oloprosencefalia, con la trisomia 13 che è la più comune, sebbene questa non sia un’associazione costante (Kallen, 1992).
Si pensa che la patogenesi implichi un difetto nei geni di segnalazione responsabili della regolazione del pattern del tubo neurale., I risultati intracranici includono ipoplasia corticale variabile, fusione variabile del diencefalo, gangli basali e talamo e presenza di una cisti dorsale (derivante da talami fusi) che si espande da un ventricolo 3rd parzialmente bloccato (Simon, 2001). L ‘idrocefalo, causato da un accumulo anomalo di liquido cerebrospinale nei ventricoli, non è raro nell’ oloprosencefalia e si pensa sia dovuto a malformazione dei ventricoli o ad eccessiva produzione di liquido cerebrospinale., Questo spesso complica la classificazione dell’oloprosencefalia, poiché il cervello è compresso e il cranio precedentemente microcefalico può espandersi prima della fusione delle suture craniche (Tripathi, 2009). È importante affrontare la visione del paziente per consentire un’interazione ottimale con l’ambiente circostante. Spesso gli occhiali non sono tollerati a causa del grado di asimmetria facciale e anomalie strutturali presenti. In queste situazioni, riteniamo che le lenti a contatto dovrebbero essere considerate come un modo per migliorare la funzione visiva.,
Sebbene vi sia una variazione dell’inserzione del muscolo retto mediale nella popolazione generale, questo caso è notevole per le inserzioni anomale del muscolo extraoculare, in particolare la distanza di 7 mm del retto mediale dal limbus. Come accennato in precedenza, i difetti della linea mediana sono comuni nell’oloprosencefalia, il che può spiegare perché i retti mediali sono stati coinvolti preferenzialmente. Con l’eccezione della ciclopia, poco è stato pubblicato sulle associazioni oculari e strabismiche con oloprosencefalia.,
Nel complesso, il trattamento è altamente individualizzato in base alla gravità del paziente e alla configurazione della malformazione. Il trattamento è di supporto e sintomatico e la prognosi dipende molto dal tipo di oloprosencefalia e dalle sue anomalie associate (Nanni, 2000).
Nel caso presentato sopra, una delle considerazioni più significative per la famiglia del paziente era come intervenire per migliorare la visione e consentire una crescita e uno sviluppo continui. Il paziente era gravemente malato nei primi anni di vita e le cure palliative sono state inizialmente discusse come un’opzione praticabile., La famiglia del paziente ha voluto continuare il trattamento e cercare possibili interventi per migliorare la qualità della vita. Di fronte a situazioni difficili come questa, è essenziale per gli operatori sanitari per aiutare le famiglie a prendere decisioni in un ambiente rispettoso senza giudizio o influenza indebita. L’American College of Critical Care Medicine Task Force ha pubblicato linee guida sulla pratica clinica che affrontano il supporto della famiglia nell’unità di terapia intensiva centrata sul paziente (Davidson, 2007)., Attraverso una buona comunicazione, gestione dei conflitti e capacità di incontro facilitazione, le famiglie possono essere coinvolti in un modello decisionale condiviso in cui le famiglie non sono gli unici responsabili di tutte le decisioni mediche autonomamente, né sono fornitori di cure paternalistiche. Durante le riunioni di famiglia, si raccomanda ai membri della famiglia di porre domande aperte sulla loro comprensione della cura del paziente, delle loro paure e delle strategie di coping., I prestatori di assistenza sono quindi incoraggiati a ripetere i sentimenti della famiglia per consentire lo sviluppo della fiducia nel team e nel processo decisionale. Dopo questo, i professionisti dovrebbero fornire informazioni chiare e oneste in un linguaggio accessibile, con l’opportunità di porre domande. L’obiettivo della discussione è il consenso, che è aiutato dal riconoscimento rispettoso di tutte le opinioni.,
Penticuff e Arheart hanno studiato l’efficacia degli incontri tra operatori sanitari e genitori in un ambiente di terapia intensiva neonatale e hanno dimostrato che il processo decisionale condiviso ha portato a meno conflitti, aspettative non realistiche dei genitori e una migliore collaborazione, oltre ad aiutare i genitori a capire meglio la situazione medica del loro bambino (Penticuff, 2005). I livelli di stress familiare sono mostrati diminuiti con una comunicazione aperta ed efficace, così come un ambiente di speranza (Davidson, 2007)., È importante sottolineare che tale cura centrata sul paziente ha dimostrato di migliorare anche i risultati clinici (Lewin, 2001).
Diagnosi Differenziale
- Esotropia
- Monoculare elevazione carenza
- Dissociata deviazione verticale
Sommario
|
Segni
|
Sintomi
|
Trattamento
|
Barkovich AJ, Simon EM, Clegg NJ, Parente SL, Hahn JS. Analisi della corteccia cerebrale in oloprosencefalia con attenzione alle fessure sylviane. ADJNR Am J Neuroradiol. 2002;23(1):143-50.
Blaas HG, Eriksson AG, Salvesen KA, Isaksen CV, Christensen B, Møllerløkken G, Eik-Nes SH., Cervelli e volti in oloprosencefalia: descrizione pre e postnatale di 30 casi. Ultrasuoni Obstet Gynecol. 2002;19(1):24-38.
Barra Jr M, Hanson JW, Currey K, Sharp S, Toriello H, Schmickel RD, Wilson GA. Oloprosencefalia nei neonati di madri diabetiche. J Pediatr 1983; 102: 565D8.
Davidson JE, Powers K, Hedayat KM, Tieszen M, Kon AA, Shepard E, Spuhler V, Todres ID, Levy M, Barr J, Ghandi R, Hirsch G, Armstrong D., Linee guida di pratica clinica per il supporto della famiglia nell’unità di terapia intensiva centrata sul paziente: American College of Critical Care Medicine Task Force 2004-2005. American College of Critical Care Medicine Task Force 2004-2005, Society of Critical Care Medicine. Crit Cura Med. 2007;35(2):605-22.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Oloprosencefalia. Orphanet J Raro Dis. 2007 2;2:8.
Kallen B, Castilla EE, Lancaster PAL, et al. Il ciclope e la sirena: uno studio epidemiologico di due tipi di malformazioni rare. J Med Genet 1992; 29: 30-35.,
Lewin SA, Skea ZC, Entwistle V, Zwarenstein M, Dick J. Interventi per i fornitori per promuovere un approccio centrato sul paziente nelle consultazioni cliniche. Cochrane Database Syst Rev. 2001; (4): CD003267. (PMID:11687181)
Munke M. Approcci clinici, citogenetici e molecolari all’eterogeneità genetica dell’oloprosencefalia. Am J Med Genet 1989; 34:237-245.
Nanni L, Schelper RL, Muenke MT. Genetica molecolare dell’oloprosencefalia. Biosci anteriore. 2000 1;5: D334-42.
Penticuff JH, Arheart KL.,Efficacia di un intervento per migliorare la collaborazione genitore-professionale in terapia intensiva neonatale. J Perinat Nurs neonatali. 2005;19(2):187-202.
Scott WE, Jackson OB. Doppia paralisi dell’elevatore: il significato della restrizione del retto inferiore. Am Orthopt J. 1977;27: 5-10.
Simon EM, Hevner RF, Pinter J, Clegg NJ, Delgado M, Kinsman SL, Hahn JS, Barkovich AJ. La cisti dorsale in oloprosencefalia e il ruolo del talamo nella sua formazione. Neuroradiologia. 2001;43(9):787-91.
Tripathi AK, Agrawal D, Sedain G. Idrocefalo oloprosencefalia: Un ossimoro?, Approfondimenti in eziologia e gestione. J Pediatr Neurosci. 2009;4(1):41-3.
Consenso per l’utilizzo di foto e video ottenuti dalla madre del paziente.
Formato di citazione suggerito: Kemp PS, Casey, G, Longmuir SQ. Oloprosencefalia e Strabismus.Eyerounds.org. 12 Giugno 2012, Disponibile da:http://EyeRounds.org/cases/151-holoprosencephaly-strabismus.htm