Quali sono le differenze nelle classi di dispositivi medici FDA?
La FDA degli Stati Uniti regola tutti i dispositivi medici commercializzati negli Stati Uniti, che sono raggruppati in tre grandi classi. Qualsiasi dispositivo medico approvato dalla FDA è classificato come classe I, II, o III a seconda del rischio del dispositivo, invasività, e l”impatto sulla salute generale del paziente. Ma dove sono le linee tracciate tra ciascuna di queste tre classi, e perché?,
Le linee guida di classificazione della FDA degli Stati Uniti possono essere molto confuse per i produttori di dispositivi medici con un’esposizione limitata al sistema. C’è un’enorme differenza nel percorso ottimale di mercato per i produttori a seconda di come il dispositivo è raggruppato. I dispositivi di classe I sono soggetti a molti meno requisiti normativi rispetto ai dispositivi di classe II o III.
Comprendendo le differenze nelle classi di dispositivi medici FDA, puoi capire come verrà raggruppato il tuo dispositivo., Con queste conoscenze in mano, i produttori di dispositivi medici nelle fasi di pre-mercato possono preparare e allocare meglio le risorse necessarie per l’approvazione normativa.
Differenze tra le classi di dispositivi medici FDA
La FDA ha classificato oltre 1.700 tipi distinti di dispositivi medici. I dispositivi sono organizzati nel Codice dei regolamenti federali (CFR) secondo le specialità 16, come i dispositivi cardiovascolari o ematologici., Classificare il tuo dispositivo medico in base a una delle 16 specialità è il primo passo per capire se stai producendo un dispositivo medico di classe I, II o III.
Dopo aver classificato un dispositivo in base alla specialità, la FDA istruisce i produttori a procedere alla notifica pre-mercato con la consapevolezza se il loro dispositivo è esente o meno. I dispositivi medici di classe I, la categoria meno rischiosa e invasiva, sono esenti dai processi di notifica pre-mercato. Anche i dispositivi specifici di classe II sono esenti dall’approvazione pre-mercato.,
Tuttavia, tutti i dispositivi regolati dalla FDA sono soggetti agli attuali requisiti di buona pratica di produzione (cGMP) per la registrazione, l’etichettatura e la qualità. Ma come fai a sapere se il dispositivo è di classe I o II, e se siete tenuti a sottoporsi a notifica premarket? 
Classe 1
La FDA degli Stati Uniti definisce i dispositivi di Classe I come dispositivi che “non sono destinati all’uso nel sostenere o sostenere la vita o di importanza sostanziale nel prevenire danni alla salute umana e potrebbero non presentare un potenziale rischio irragionevole di malattia o lesioni.,”
Questi dispositivi sono la classe più comune di dispositivi regolamentati dalla FDA, che costituiscono il 47 per cento dei dispositivi approvati sul mercato.
I dispositivi di classe I hanno un contatto minimo con i pazienti e un basso impatto sulla salute generale del paziente. In generale, i dispositivi di classe I non entrano in contatto con gli organi interni del paziente, il sistema nervoso centrale o il sistema cardiovascolare. Questi dispositivi sono soggetti al minor numero di requisiti normativi.,
Esempi di Dispositivi di Classe I:
- Spazzolino da denti Elettrico
- Lingua Depressore
- la Maschera di Ossigeno
- Riutilizzabili Bisturi
- Bende
- Letti di Ospedale
Portare Dispositivi Medici di Classe I di Mercato
Dispositivi di classe I sono il più semplice e veloce per portare sul mercato poiché la quantità minima di rischio per il paziente e sono raramente fondamentale per sostenere la sua vita per la cura. La maggior parte dei dispositivi di classe I sono esenti dai requisiti FDA per la notifica Premarket (510k) e l’approvazione Premarket (PMA).,
I dispositivi di classe I non sono esenti dai controlli generali FDA, una serie di comandi che si applica ai dispositivi medici di classe I, II e III. Le disposizioni di questa legge riguardano adulterazione, misbranding, registrazione dei dispositivi, record e buone pratiche di produzione. I produttori di dispositivi medici che rientrano nella classe A devono ancora implementare un sistema di gestione della qualità e seguire gli standard per garantire un prodotto di qualità.,
LETTURA CORRELATA: La differenza tra la notifica Premarket 510 (k) e l’approvazione Premarket
Classe 2
I dispositivi medici di classe II sono più complicati dei dispositivi di Classe I e presentano una categoria di rischio più elevata perché hanno maggiori probabilità di entrare in contatto prolungato con un paziente. Questo può includere dispositivi che entrano in contatto con il sistema cardiovascolare o gli organi interni di un paziente, e strumenti diagnostici.,
La FDA definisce i dispositivi di Classe II come “dispositivi per i quali i controlli generali non sono sufficienti a fornire una ragionevole garanzia della sicurezza e dell’efficacia del dispositivo.,est Kit
Portare Dispositivi Medici di Classe II di Mercato
Controlli variano a seconda del dispositivo, ma secondo la FDA, possono includere:
- le prestazioni del Dispositivo
- Postmarket di sorveglianza
- registri di Pazienti
- Particolari requisiti di etichettatura
- Premarket requisiti in materia di dati
- Linee guida
La maggior parte dei dispositivi di Classe II sono approvati dalla FDA per il mercato, attraverso il Premarket di Notifica, o 510(k) del processo.,
I dispositivi di classe II sono soggetti agli stessi controlli generali sopra menzionati, ma la FDA li definisce come “dispositivi per i quali i controlli generali non sono sufficienti a fornire una ragionevole garanzia della sicurezza e dell’efficacia del dispositivo.” Per questo motivo, i dispositivi di classe II sono anche soggetti a controlli speciali. Questi regolamenti dipendono dal dispositivo e possono includere requisiti di etichettatura speciali, registri dei pazienti e standard di prestazioni.
La maggior parte dei dispositivi di Classe II arriva sul mercato utilizzando il processo di notifica Premarket (510k)., Il 510 (k) è un’applicazione complessa per la FDA, che dimostra che un dispositivo è sicuro ed efficace dimostrando che il dispositivo è equivalente a un altro dispositivo che è sul mercato.
Questo processo comporta la visualizzazione di “equivalenza sostanziale” a un altro dispositivo che è noto nel linguaggio FDA come “il predicato.”Questo non significa che i dispositivi devono essere identici, ma richiedono somiglianze significative in uso, design, materiali, etichettatura, standard, e altre caratteristiche.,
La FDA ha rilasciato una lista di esenzione all’inizio del 2018 che esenta oltre 800 dispositivi medici generici di classe I e II dal processo 510(k). Se si dispone di un dispositivo medico generico di classe II, è possibile scoprire se è esente da un deposito di 510(k) cercando nel database di classificazione dei prodotti FDA.
LETTURA CORRELATA: 5 Motivi Revisione FDA 510 (k) è una grande mossa.
Classe 3
La FDA definisce i dispositivi di Classe III come prodotti che “di solito sostengono o sostengono la vita, sono impiantati o presentano un potenziale rischio irragionevole di malattia o lesioni.,”
Solo il 10 per cento dei dispositivi regolamentati dalla FDA degli Stati Uniti rientrano nella classe III. Questa classificazione è generalmente estesa a impianti permanenti, dispositivi medici intelligenti e sistemi di supporto vitale.
Mentre la classe III è generalmente riservata ai dispositivi medici più innovativi e all’avanguardia, ci sono altri dispositivi che possono rientrare nella Classe III per diversi motivi., Alcuni dispositivi inizialmente classificati come Classe II possono essere spostati fino alla classe III se il produttore non è in grado di dimostrare l’equivalenza sostanziale a un predicato (prodotto esistente) durante il processo di archiviazione PMA (510k).,
Esempi di Dispositivi Medici di Classe III:
- protesi al Seno
- Pacemaker
- Defibrillatori
- Alta frequenza ventilatori
- gli impianti Cocleari
- il prelievo di sangue Fetale monitor
- Impiantato protesi
Portando Dispositivi Medici di Classe III di Mercato
Classe III dispositivi sono soggetti a tutte FDA Controlli di carattere Generale e la FDA Premarket di Approvazione (PMA) del processo., La FDA scrive: “a causa del livello di rischio associato ai dispositivi di classe III, la FDA ha stabilito che i controlli generali e speciali da soli non sono sufficienti per garantire la sicurezza e l’efficacia dei dispositivi di classe III.”
Il PMA è il tipo più intensivo di applicazione di marketing del dispositivo richiesto dalla FDA. Alcuni dispositivi FDA Classe III sono esenti e possono beneficiare di un 510 (k) deposito, ma la maggior parte si prevede di ottenere l’approvazione Premarket.,
Il processo PMA richiede uno studio rigoroso di un dispositivo medico per dimostrare la sicurezza e l’efficacia attraverso lo sviluppo di un profilo di rischio/beneficio basato sui dati. Il processo PMA comporta generalmente studi clinici e tempo e risorse significativi per una raccolta di dati sufficiente. Le uniche eccezioni al processo PMA all’interno della classe III sono dispositivi con un equivalente sostanziale. È possibile determinare se un dispositivo di classe III può essere commercializzato con un 510 (k) cercando nel database di approvazione Premarket FDA (PMA) e nel database di notifica Premarket 510(k).,
Come determinare la classe
Il primo passo verso la classificazione del dispositivo medico è quello di navigare il regolamento di classificazione FDA, l’elenco di 16 categorie per i dispositivi medici in base alla specializzazione medica.
Ad esempio, vi mostreremo i passaggi per identificare la classificazione di un allarme di pressione sanguigna. Il dispositivo è classificato nella categoria 870: Dispositivi cardiovascolari.,
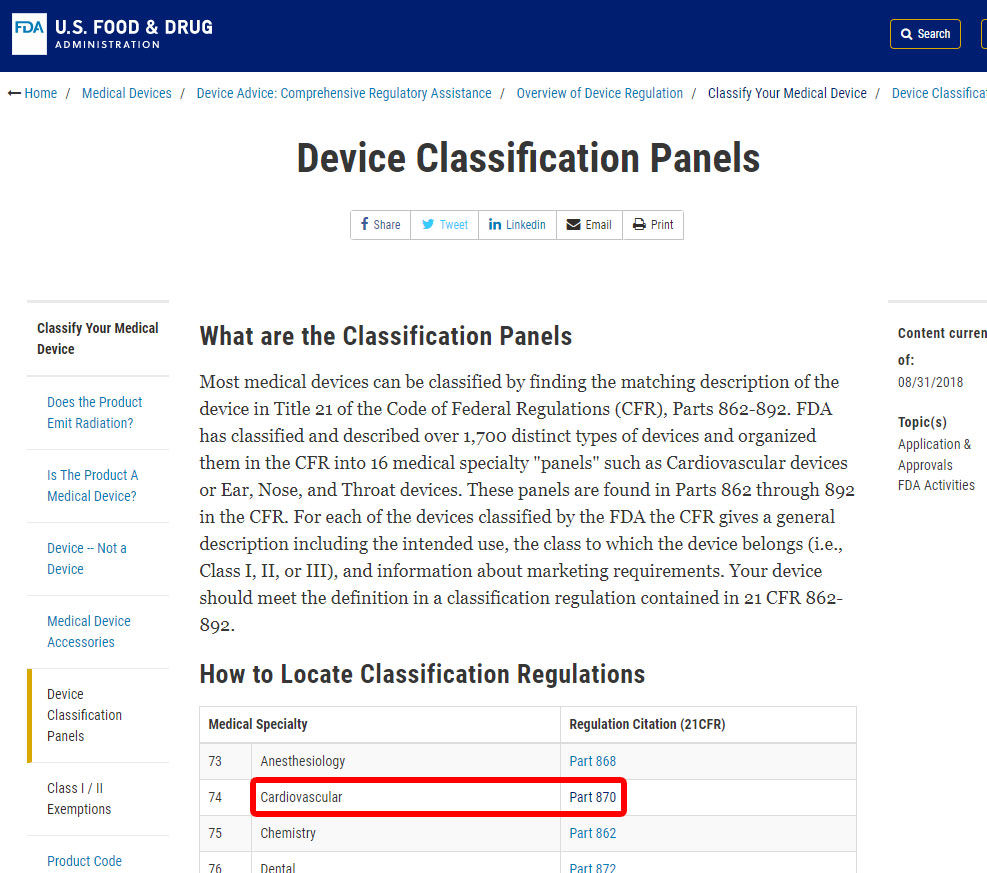
Una volta individuata la specialità medica pertinente, fare clic sulla categoria e navigare nell’elenco dei dispositivi fino a trovare un equivalente e il codice del dispositivo associato.
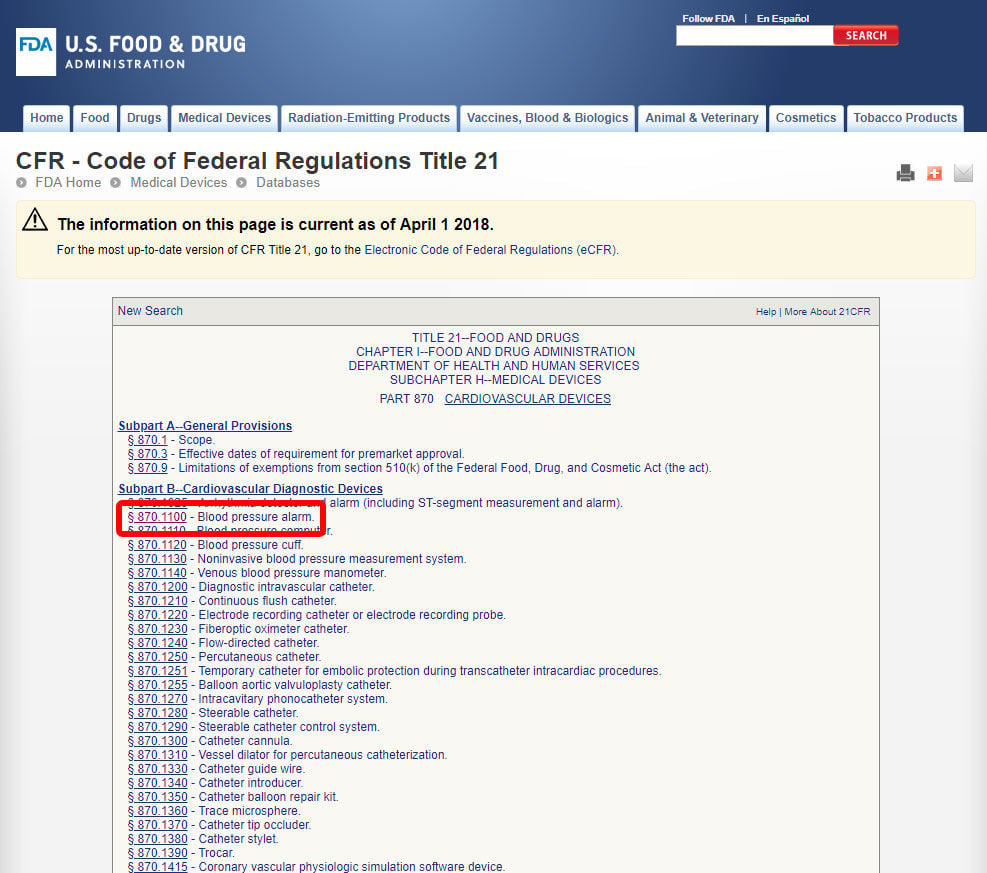
Fare clic sul codice del dispositivo e aprire le linee guida. La classificazione del dispositivo è elencata alla sezione (b).,
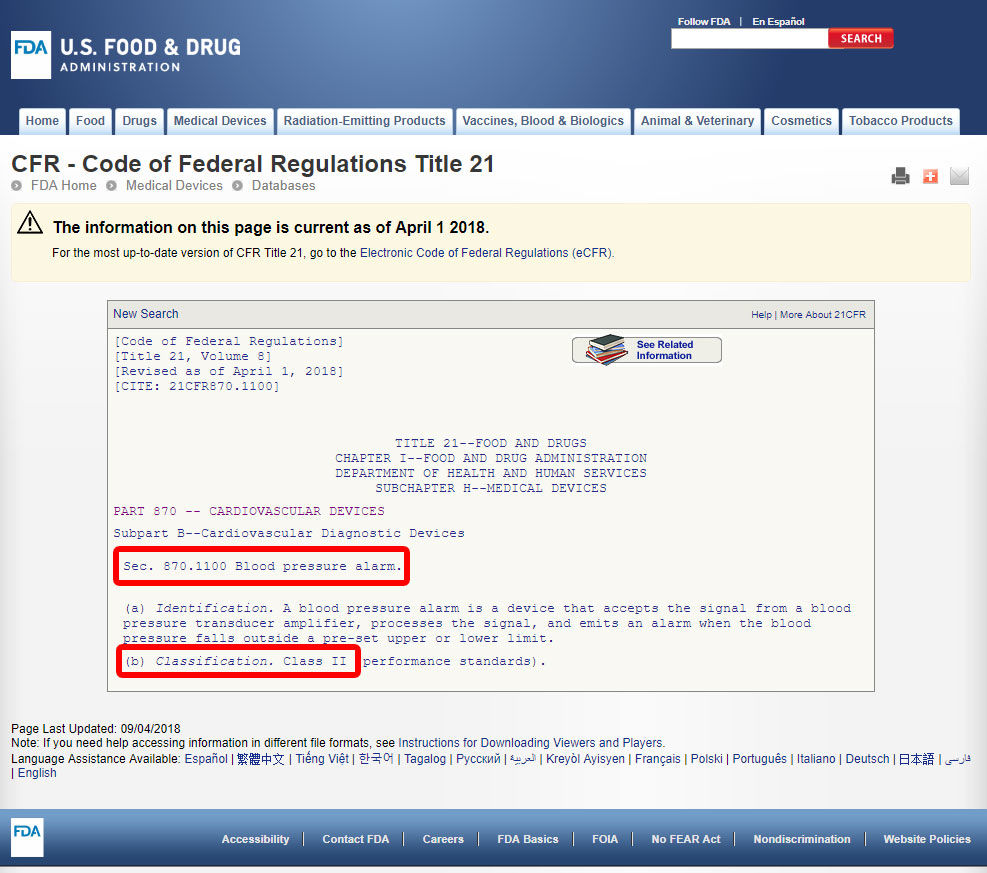
Se il vostro dispositivo non dispone di una elencate equivalente tra i 1.700 dispositivi classificati dalla FDA, è più probabile un dispositivo innovativo senza un sostanziale equivalente, e che potrebbe essere classificato come Classe III.
la Comprensione FDA Dispositivo Medico Classi
Le differenze tra i classificati come dispositivi medici di Classe I, II, o III da parte della FDA è in gran parte il rischio, la quantità di contatto con il malato e i loro sistemi interni, e se un dispositivo è fondamentale per sostenere la vita.,
Oltre a questi fattori, la FDA considera l’equivalenza sostanziale nel determinare come un dispositivo è classificato. Se il tuo dispositivo è a basso rischio e contatta il paziente in modo minimo, è probabile che tu possa qualificarti per la Classe I e un processo di approvazione del mercato semplificato. I dispositivi di classe II e III devono dimostrare la sicurezza tramite l’equivalenza sostanziale, un deposito di 510(k) o il processo di approvazione pre-mercato.
Sapendo come il dispositivo è classificato, è possibile semplificare il percorso di approvazione del mercato comprendendo i processi e documenti che sono suscettibili di essere richiesto dalla FDA., Se la tua organizzazione è soggetta a un requisito di Classe II o Classe III 510(k) o PMA, questa conoscenza può aiutarti ad allocare le risorse appropriate in anticipo e pianificare un deposito di successo.
