FDA医療機器クラスの違いは何ですか?
米国FDAは、米国で販売されているすべての医療機器を規制しています。 FDAによって承認された医療機器は、装置のリスク、侵襲性、および患者の全体的な健康への影響に応じて、クラスI、II、またはIIIのいずれかに分類され しかし、これら三つのクラスのそれぞ,
米国FDAの分類ガイドラインは、システムへの暴露が限られている医療機器製造業者にとって非常に混乱する可能性があります。 が大きな差を最適なパスを市場メーカーによってご利用を停止してください。グループ化します。 クラスi機器の対象というのは、ごく限られた規制要件によりクラスII及びIIIです。
FDA医療機器クラスの違いを理解することにより、デバイスがどのようにグループ化されるかを理解することができます。, この知識を手に入れることで、市場前段階の医療機器メーカーは、規制当局の承認に必要なリソースをより適切に準備し、割り当てることができます。
FDA医療機器クラスの違い
FDAは1,700以上の異なるタイプの医療機器を分類しています。 これらのデバイスは、心臓血管または血液学デバイスなどの16の専門分野に従って、連邦規則(CFR)のコードで編成されています。, 16の専門分野のいずれかに従って医療機器を分類することは、クラスI、II、またはIII医療機器を製造しているかどうかを理解するための最初のス
専門に従って装置を分類した後、FDAは製造業者に装置が免除されているかどうかの知識の市販前通知に進むように指示する。 最も危険で侵襲的なカテゴリーであるクラスI医療機器は、市販前通知プロセスから免除されます。 特定のクラスIIデバイスも市販前承認から免除されます。,ただし、FDAによって規制されているすべてのデバイスは、登録、ラベリング、および品質に関する現在のGood Manufacturing Practice(cGMP)要件の対象となります。 がどのようにして知りましたかの場合はクラスからなる”かねますが、希望する場合は申請によpremarket通知? 
クラス1
米国FDAは、クラスIデバイスを”生命を支えたり維持したりするための使用を意図していないか、または人間の健康に対する障害を防ぐためにかなり重要なデバイスと定義しており、病気やけがの潜在的な不合理なリスクを示さない可能性がある。,”
これらのデバイスは、FDAによって規制されている最も一般的なクラスのデバイスであり、市場で承認されたデバイスの47%を構成しています。
クラスI装置に患者が付いている最低の接触および患者の全面的な健康の低い影響があります。 一般に、クラスi装置は、患者の内臓、中枢神経系、または心臓血管系と接触しない。 これらのデバイスは、規制要件が最も少なくなります。,
クラスIデバイスの例:
- 電動歯ブラシ
- 舌圧子
- 酸素マスク
- 再利用可能な外科用メス
- 包帯
- 病院ベッド
クラスI医療機器を市場に持ち込む
クラスIデバイスは、患者に最も低いリスク量を示し、まれにしか市場に持ち込まれないため、市場に持ち込むのが最も速く、最も簡単です。生命維持ケアに不可欠です。 クラスIデバイスの大部分は、市販前通知(510k)および市販前承認(PMA)に関するFDA要件を免除されています。,クラスIデバイスは、クラスI、II、およびIIIの医療機器に適用される一連のコマンドであるFDA general controlから免除されません。 この法律の規定は、混入、誤ブランディング、デバイス登録、記録、および適正製造慣行に対処しています。 クラスAに該当する医療機器メーカーは、依然として品質マネジメントシステムを実装し、高品質の製品を確保するための基準に従う必要があります。,
関連読書:市販前通知510(k)と市販前承認の違い
クラス2
クラスII医療機器は、クラスI機器よりも複雑であり、患者と持続的に接触する可能性が高いため、リスクのカテゴリーが高くなります。 これには、患者の心臓血管系または内臓と接触する装置、および診断ツールが含まれ得る。,
FDAは、クラスIIデバイスを”一般的な制御がデバイスの安全性と有効性の合理的な保証を提供するには不十分なデバイス”と定義しています。,estキット
クラスII医療機器を市場に出す
コントロールは、デバイスによって異なりますが、FDAによると、以下を含むことができます。
- デバイスのパフォーマンス
- 市販後の監視
- 患者レジストリ
- 豊胸インプラント
- ペースメーカー
- 除細動器
- 高周波換気装置
- 人工内耳
- 胎児の血液サンプリングモニター
- 埋め込まれた補綴
クラスiiデバイスの大部分は、市販前通知、または510(k)プロセスを通じて市場で承認されたfdaです。,
クラスIIデバイスは、上記と同じ一般的なコントロールの対象となりますが、FDAは、それらを”一般的なコントロールがデバイスの安全性と有効性の合理的な保証を提供するには不十分であるデバイスであると定義しています。”そのため、クラスIIデバイスも特別な制御の対象となります。 これらの規則は装置によって決まり、特別な分類の条件、忍耐強い記録および標準作業時間を含むかもしれない。
ほとんどのクラスIIデバイスは、市販前通知(510k)プロセスを使用して市場に出てきます。, 510(k)はFDAへの複雑な適用であり、装置が市場にある別の装置と同等であることを示すことによって装置が安全、有効であることを示す。
このプロセスは、FDAの用語で”述語”として知られている別のデバイスに”実質的な等価性”を示すことを含む。”これは、デバイスが同一である必要があることを意味しませんが、使用、設計、材料、ラベル、規格、およびその他の特性において重要な類似点が必要です。,
FDAは2018年初めに、800以上の一般的なクラスIおよびII医療機器を510(k)プロセスから免除する免除リストを発表しました。 一般的なクラスII医療機器をお持ちの場合は、FDA製品分類データベースを検索することで、510(k)申請が免除されているかどうかを確認できます。
関連読書:5つの理由FDA510(k)をオーバーホールすることは大きな動きです。
クラス3
FDAは、クラスIIIデバイスを”通常、生命を維持またはサポートし、移植され、または病気または怪我の潜在的な不合理なリスクを提示する製品と,”
米国FDAによって規制されているデバイスのわずか10パーセントは、クラスIIIに分類されます。
クラスIIIは一般的に最も革新的で最先端の医療機器のために予約されていますが、さまざまな理由でクラスIIIに分類できる他のデバイスが, 最初にクラスIIとして分類されるある装置は製造業者がpma(510k)ファイリングプロセスの間に述語(既存のプロダクト)に相当な等価性を示してなければクラスIIIまでぶつかるかもしれません。,
クラスIII医療機器の例:
クラスIII医療機器を市場に投入する
クラスIII機器は、fda一般管理およびfda市販前承認(Pma)プロセス。, FDAは、”クラスIIIデバイスに関連するリスクのレベルのために、FDAは、一般的および特別なコントロールだけでは、クラスIIIデバイスの安全性および有効性を保証するには不十分であると判断しました。”
PMAは、FDAによって要求される最も集中的なタイプのデバイスマーケティングアプリケーションです。 一部のFDAクラスIIIデバイスは免除されており、510(k)申請の対象となる場合がありますが、大部分は市販前の承認を得ることが期待されています。,
PMAプロセスでは、データ駆動型の利益/リスクプロファイルの開発を通じて安全性と有効性を証明するために、医療機器の厳格な研究が必要です。 PMAプロセスは、一般に、臨床試験および十分なデータ収集のためのかなりの時間および資源を含む。 クラスIII内のPMAプロセスの唯一の例外は、実質的に同等のデバイスです。 FDA市販前承認(PMA)データベースおよび510(k)市販前通知データベースを検索することにより、クラスIIIデバイスを510(k)で販売できるかどうかを判断できます。,
あなたのクラスを決定する方法
あなたの医療機器を分類するための最初のステップは、FDAの分類規則、医療専門に応じた医療機器の16のカテゴリのリストをナビゲートすることです。
例として、血圧アラームの分類を特定する手順を示します。 装置は部門870の下で分類される:心血管装置。,
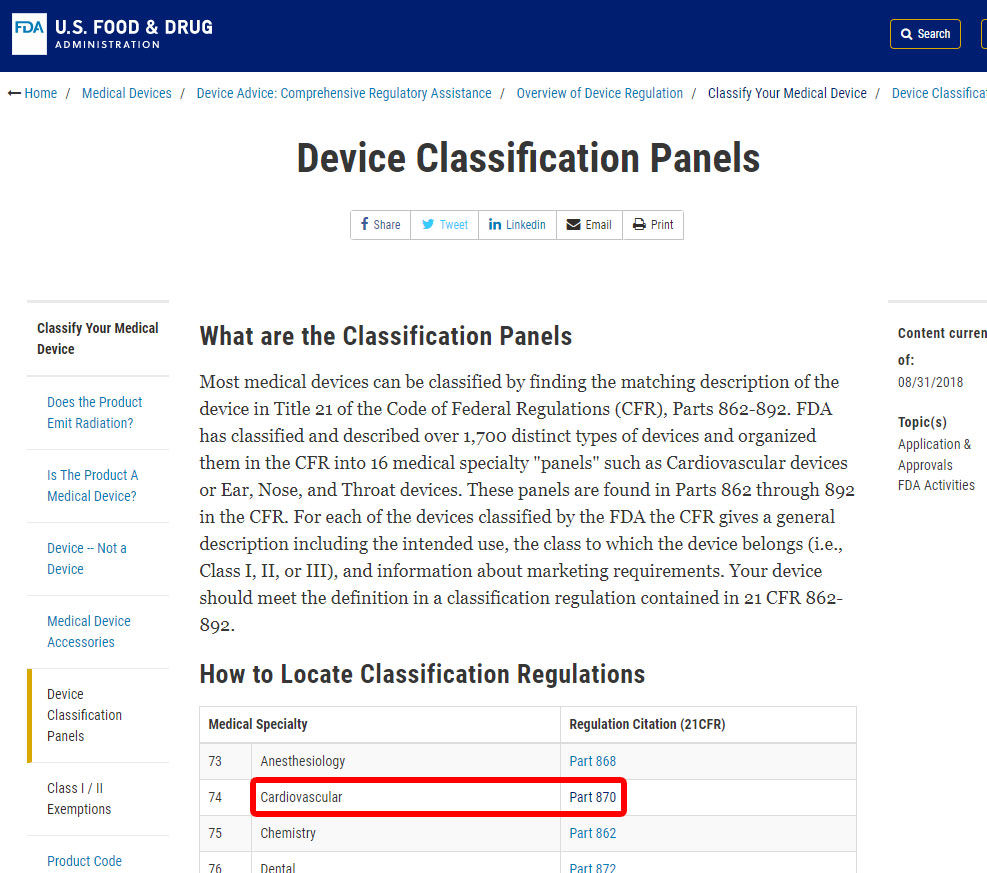
関連する医療専門分野を見つけたら、カテゴリをクリックして、同等のデバイスと関連するデバイスコードが見つかるまでデバイスのリストをナビゲートします。
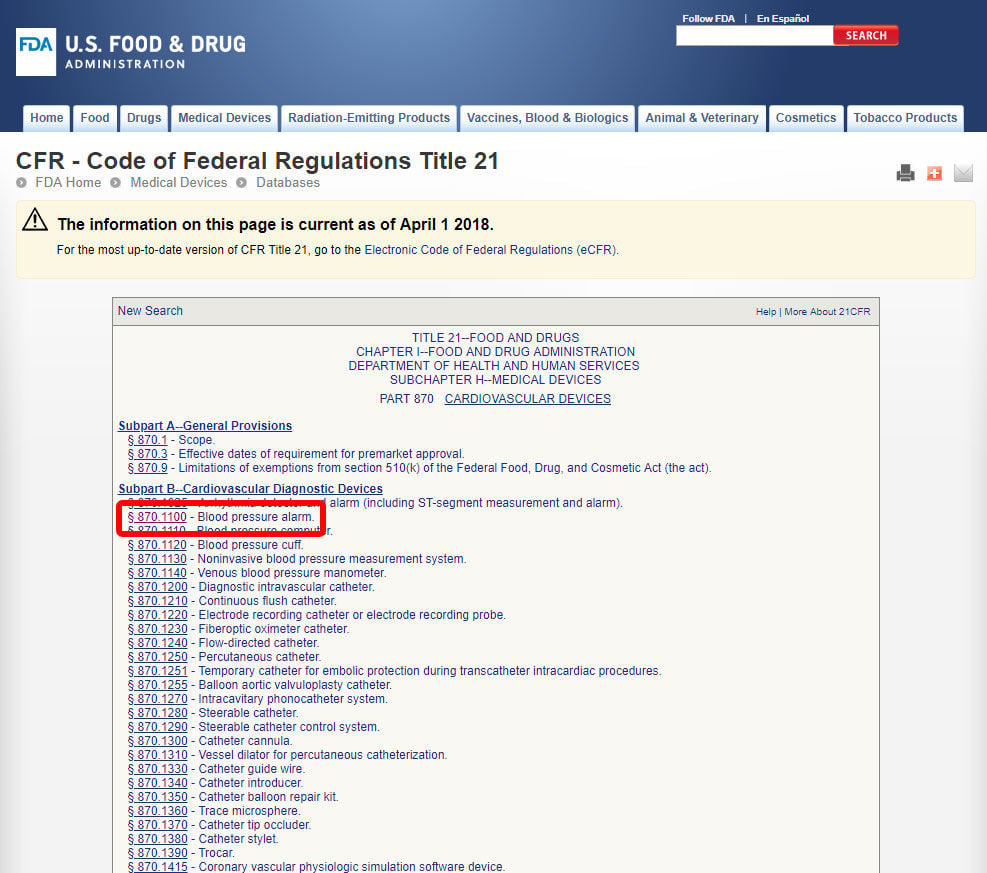
デバイスコードをクリックしてガイドラインを開きます。 デバイスの分類は、セクション(b)に記載されています。,
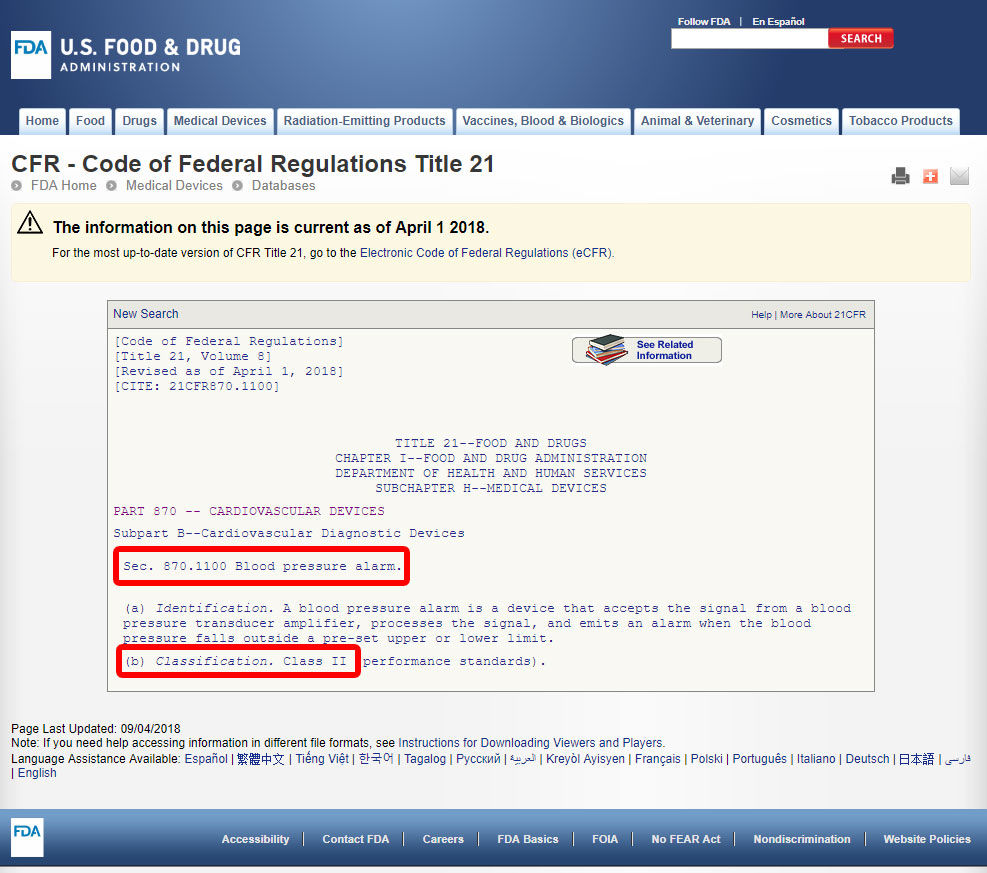
お使いのデバイスにFDAによって分類された1,700個のデバイスの中で同等のものがない場合、実質的な同等のものがない革新的なデバイスであり、クラスIIIに分類される可能性が最も高い。
FDA医療機器クラスの理解
FDAによってクラスI、II、またはIIIに分類される医療機器の違いは、主にリスク、患者との接触量およびその内部システム、およびデバイスがクラスIIIに分類されるかどうかである。生命を支えることに重大。,
これらの要因に加えて、FDAは、デバイスがどのように分類されるかを決定する際に実質的な等価性を考慮する。 お使いのデバイスが低リスクで、患者と最小限に接触する場合は、クラスIおよび合理化された市場承認プロセスの対象となる可能性があります。 クラスIIおよびIIIデバイスは、実質的な同等性、510(k)申請、または市販前承認プロセスを通じて安全性を実証する必要があります。
お使いのデバイスがどのように分類されているかを知ることで、FDAによって要求される可能性のあるプロセスと文書を理解することで、市場承認, 組織がクラスIIまたはクラスIII510(k)またはPMA要件の対象となっている場合、この知識は、適切なリソースを事前に割り当て、申請の成功を計画するのに
