Hva Er Forskjellene i FDA Medisinsk Enhet Klasser?
Den AMERIKANSKE FDA regulerer alt medisinsk utstyr som er markedsført i USA, som er gruppert i tre brede klasser. Noen medisinsk enhet godkjent av FDA er klassifisert som Klasse i, II, eller III, avhengig av enheten»s risiko, invasivitet, og innvirkning på pasientens»s generelle helse. Men hvor er de linjer trukket mellom hver av disse tre klassene, og hvorfor?,
Den AMERIKANSKE FDA»s klassifisering retningslinjer kan være svært forvirrende for medisinsk utstyr produsenter med begrenset eksponering til systemet. Det er en enorm forskjell på optimal vei til markedet for produsenter avhengig av hvordan enheten er gruppert. Klasse i-utstyr er underlagt langt færre regulatoriske krav enn Klasse II-eller III-enheter.
Ved å forstå forskjeller i FDA medisinsk enhet klasser, kan du forstå hvordan enheten vil være gruppert., Med denne kunnskapen i hånden, medisinsk utstyr produsenter i premarket stadier kan bedre forberede og fordele de ressurser som trengs for regulatorisk godkjennelse.
Forskjeller mellom FDA Medisinsk Enhet Klasser
FDA har klassifisert over 1700 forskjellige typer av medisinsk utstyr. Enhetene er organisert i Code of Federal Regulations (CFR) i henhold til 16 spesialiteter, slik som Hjerte-eller Hematologi enheter., Klassifisering av det medisinske utstyret i henhold til én av de 16 spesialiteter er første skritt for å forstå om du er produksjon av en Klasse i, II, eller III medisinske utstyret.
Etter å klassifisere en enhet i henhold til spesialitet, FDA instruerer produsenter for å gå videre til premarket varsling med kunnskap om deres enhet er fritatt eller ikke. Klasse i medisinsk utstyr, den minst risikable og invasive kategori, er unntatt fra premarket varsling. Bestemt klasse II-utstyr er også unntatt fra premarket godkjenning.,
Imidlertid alle enheter som er regulert av FDA er underlagt current Good Manufacturing Practice (cGMP) krav til registrering, merking og kvalitet. Men hvordan vet du om enheten er i Klasse i eller II, og om du er pålagt å gjennomgå premarket varsling? 
Klasse 1
Den AMERIKANSKE FDA definerer Klasse i-enheter som enheter som er «ikke ment for bruk i støtte eller opprettholde livet eller av vesentlig betydning for å hindre svekkelse av helse, og de kan ikke presentere en potensiell urimelig risiko for sykdom eller skade.,»
Disse enhetene som er mest vanlig Klasse av enheter regulert av FDA, som utgjør 47 prosent av godkjente enheter på markedet.
Klasse i-enheter har minimal kontakt med pasienter, og med lite påvirkning på en pasient»s generelle helse. Generelt, Klasse i-enheter må ikke komme i kontakt med en pasient»s indre organer, sentrale nervesystemet, eller hjerte-og karsystemet. Disse enhetene er underlagt færrest regulatoriske krav.,
Eksempler av Klasse i-Enheter:
- Elektrisk Tannbørste
- Tungen Depressor
- Oksygen Maske
- Gjenbrukbare Kirurgiske Skalpell
- Bandasjer
- Sykehus Senger
å Bringe Klasse i Medisinsk Utstyr til Markedet
Klasse i-enheter er den raskeste og enkleste å bringe til markedet siden de presenterer det laveste beløpet av risiko for pasienten og er sjelden avgjørende for livreddende behandling. De fleste av Klasse i-enheter som er unntatt fra FDA krav til Premarket Varsling (510k) og Premarket Godkjenning (PMA).,
Klasse i-utstyr er ikke fritatt fra FDA generelle kontroller, en serie med kommandoer som gjelder for Klasse i, II, og III medisinsk utstyr. Bestemmelsene i denne lov adresse adulteration, misbranding, enhet for registrering, dokumentasjon, og (good manufacturing practices). Medisinsk utstyr produsenter som faller inn i Klasse A er fortsatt nødvendig å implementere et system for kvalitetsstyring og følge standarder for å sikre en kvalitet på produktet.,
i SLEKT å LESE: Forskjellen Mellom Premarket Varsling 510(k) og Premarket Godkjenning
Klasse 2
Klasse II medisinsk utstyr er mer komplisert enn Klasse i enheter og presentere en høyere kategori av risiko, fordi de er mer sannsynlig å komme inn vedvarende kontakt med en pasient. Dette kan omfatte enheter som kommer i kontakt med en pasient»s kardiovaskulære systemet eller indre organer, og diagnostiske verktøy.,
FDA definerer Klasse II-enheter som «enheter som generelle kontroller er ikke nok for å gi rimelig grad av sikkerhet for at sikkerheten og effektiviteten av enheten.,est Kits
å Bringe Medisinsk Klasse II-Utstyr på Markedet
Kontroller variere avhengig av enheten, men i henhold til FDA, kan omfatte:
- Enhetens ytelse
- Postmarket overvåking
- Pasient register
- Spesiell merking krav
- Premarket data krav
- Retningslinjer
flertallet av Klasse II-utstyr er FDA godkjent for markedet gjennom Premarket Varsling, eller 510(k) prosess.,
Klasse II-utstyr er underlagt de samme Generelle Kontroller som er nevnt ovenfor, men FDA definerer dem som «enheter som generelle kontroller er ikke nok for å gi rimelig grad av sikkerhet for at sikkerheten og effektiviteten av enheten.»For at grunnen, Klasse II-utstyr er underlagt Spesielle Kontroller. Disse forskrifter, avhenger av enheten, og kan inneholde spesielle krav til merking, pasient register, og ytelse standarder.
de Fleste Klasse II-enheter kommer til markedet med Premarket Varsling (510k) prosess., Den 510(k) er et komplekst program til FDA, som viser at en enhet er sikker og effektiv ved å vise at enheten er ekvivalent til en annen enhet som er på markedet.
Denne prosessen innebærer å vise «betydelig ekvivalens» til en annen enhet som er kjent i FDA språkbruk som «predikatet.»Dette doesn»t bety enheter må være identiske, men de krever betydelige likheter i bruk, design, materialer, merking, standarder og andre egenskaper.,
FDA utgitt et unntak liste tidlig i 2018, som unntar over 800 generisk Klasse i og II medisinsk utstyr fra 510(k) prosess. Hvis du har en generisk Klasse II medisinske implantatet, for du kan oppdage om det er unntatt fra 510(k) innlevering ved å søke FDA Produkt Klassifisering database.
i SLEKT å LESE: 5 Grunner til å Overhale FDA 510(k) er en Stor Flytte.
Klasse 3
FDA definerer Klasse III-enheter, samt produkter som «vanligvis opprettholde eller støtte liv, er implantert eller presentere en potensiell urimelig risiko for sykdom eller skade.,»
Bare 10 prosent av enhetene som er regulert av den AMERIKANSKE FDA falle i Klasse III. Denne klassifiseringen er generelt utvidet til permanente implantater, smart medisinsk utstyr, og livsoppholdende systemer.
Mens Klasse III er vanligvis reservert for de mest innovative og banebrytende medisinsk utstyr, det er andre enheter som kan falle i Klasse III for forskjellige grunner., Noen enheter som er kategorisert i utgangspunktet som Klasse II kan være støtdempende opp til Klasse III dersom produsenten ikke er i stand til å demonstrere betydelig ekvivalens til et predikat (eksisterende produkt) under PMA (510k) innlevering prosessen.,
Eksempler på Klasse III Medisinsk Utstyr:
- Bryst implantater
- Pacemakere
- Defibrillatorer
- Høy-frekvens vifter
- cochleaimplantat
- Fosterets blod prøvetaking skjermer
- Implantat protetikk
Bringe Klasse III Medisinsk Utstyr til Markedet
Klasse III-enheter er underlagt alle FDA Generelle Kontroller og FDA Premarket Godkjenning (PMA) prosess., FDA skriver: «på grunn av nivået av risiko forbundet med Klasse III enheter, FDA har bestemt at de generelle og spesielle kontroller alene er ikke nok til å sikre sikkerheten og effektiviteten av Klasse III-enheter.»
PMA er den mest intensive type enhet markedsføring programmet som kreves av FDA. Noen FDA Klasse III enheter er unntatt og kan kvalifisere for en 510(k) innlevering, men de aller fleste hadde forventet å få Premarket godkjenning.,
PMA prosessen krever et grundig studium av en medisinsk enhet for å bevise sikkerhet og effektivitet gjennom utvikling av en data-drevet nytte/risiko-profil. Den PMA prosessen innebærer generelt kliniske studier og betydelig tid og ressurser for tilstrekkelige data collection. De eneste unntakene til PMA prosess i Klasse III er enheter med en betydelig tilsvarende. Du kan finne ut om en Klasse III-enheten, kan markedsføres med en 510(k) ved å søke FDA Premarket Godkjenning (PMA) database og 510(k) Premarket Melding database.,
Hvordan du skal Finne Din Klasse
Det første steget mot å klassifisere din Medisinske Enheten for å navigere FDA Klassifisering forskrifter, listen over 16 kategorier for medisinsk utstyr i henhold til medisinsk spesialisering.
Som et eksempel, vi vil vise deg trinn for å identifisere klassifisering av blodtrykk alarm. Enheten er klassifisert under kategorien 870: Hjerte-enheter.,
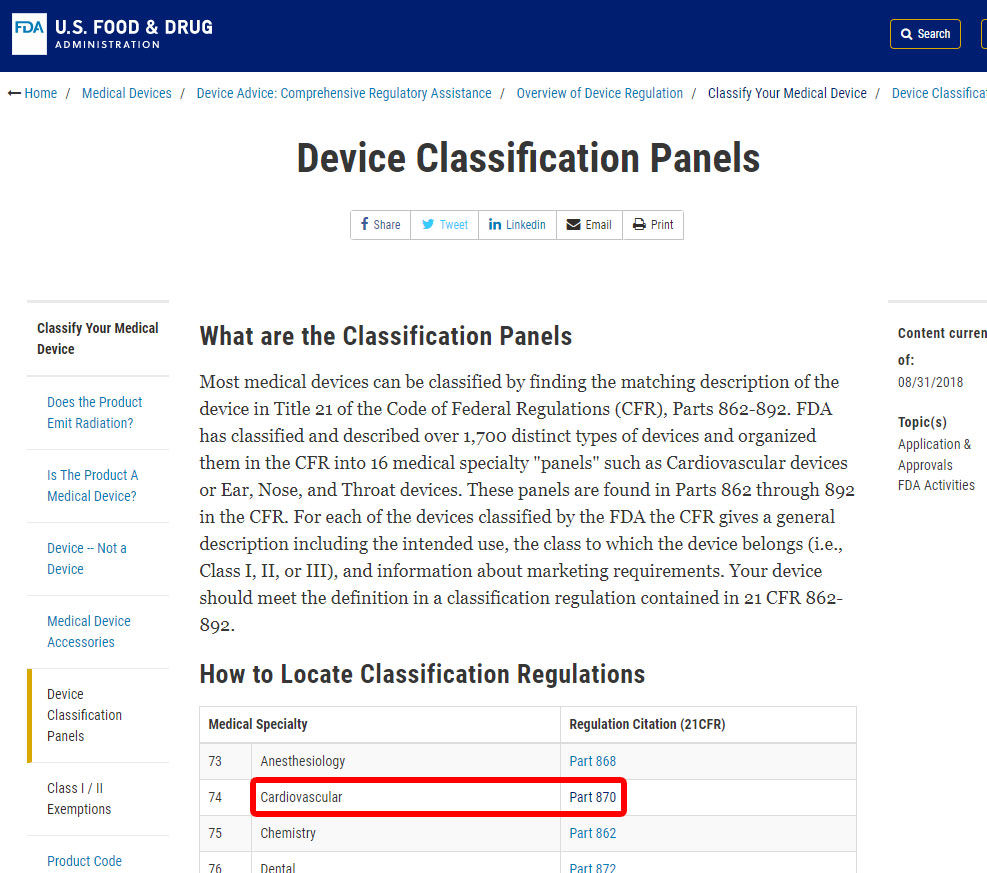
Når du»ve ligger relevant medisinsk spesialitet, klikk på kategorien, og navigere i listen over enheter til du finner et tilsvarende og den tilknyttede enheten kode.
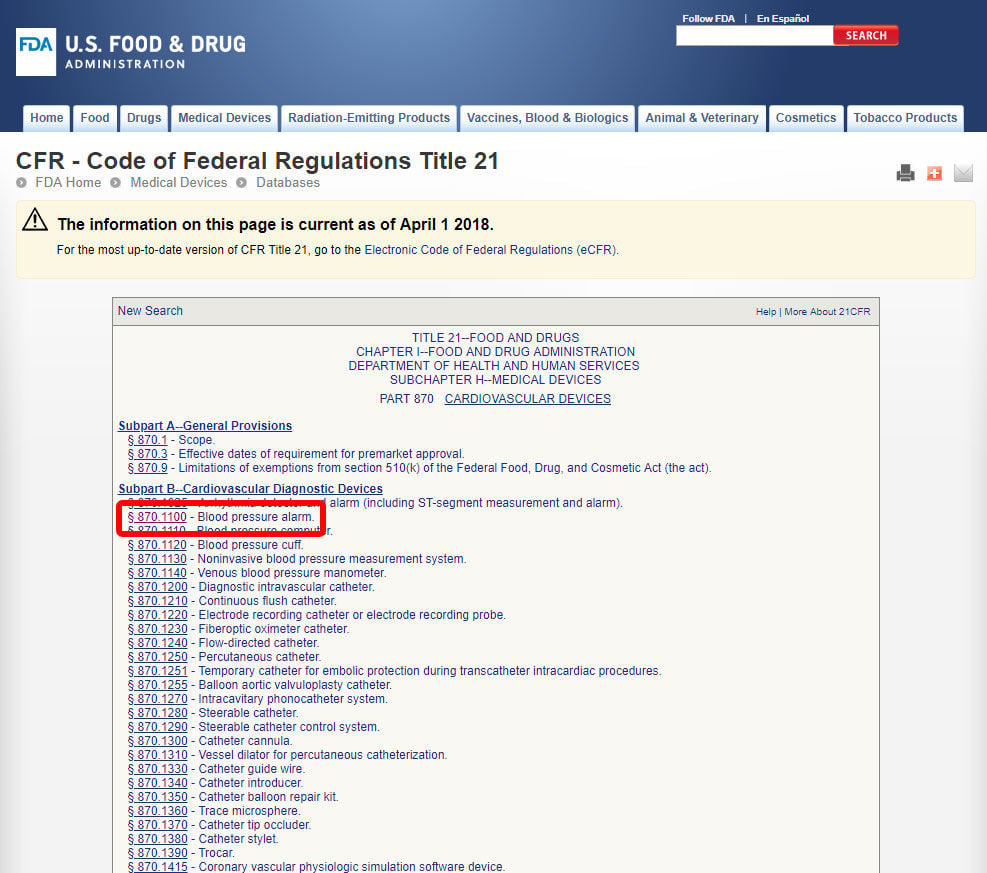
Klikk på enheten kode og åpne retningslinjer. Enheten klassifisering er oppført under punkt (b).,
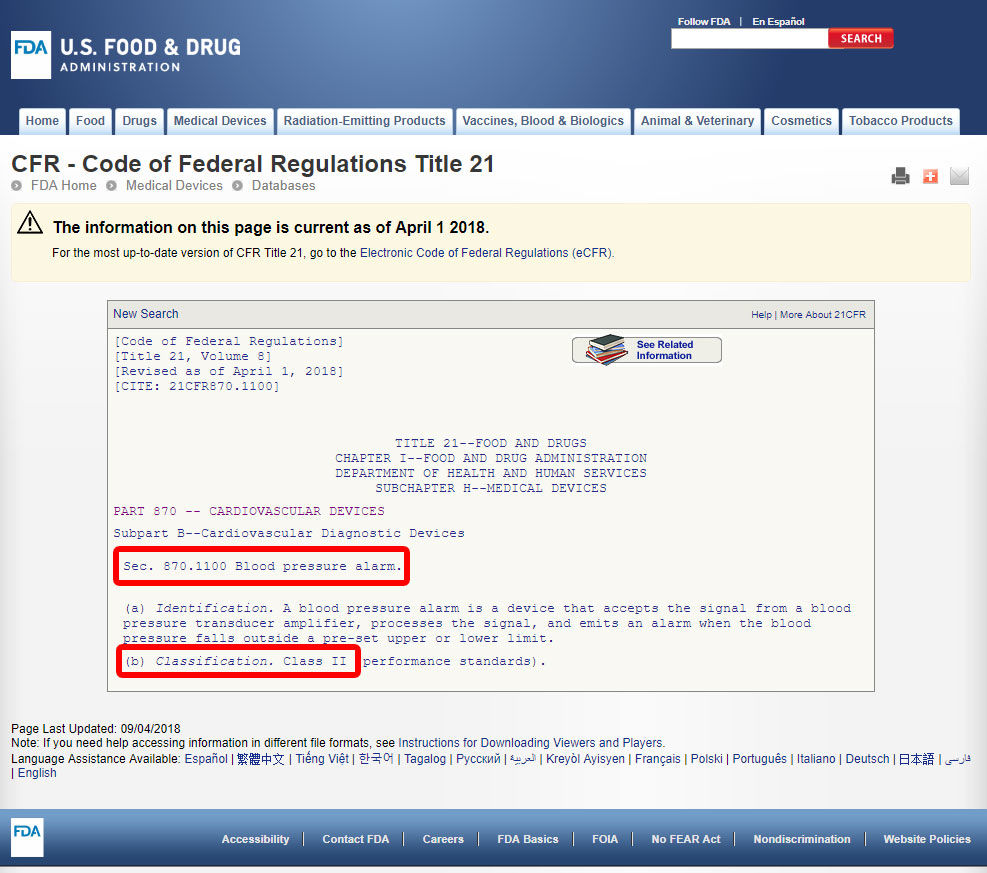
Hvis enheten din ikke er oppført tilsvarende blant 1,700 enheter klassifisert av FDA, er det mest sannsynlig en innovativ enhet uten en vesentlig tilsvarende, og vil bli klassifisert som Klasse III.
Forstå FDA Medisinsk Enhet Klasser
forskjellene mellom medisinsk utstyr som er klassifisert som Klasse i, II, eller III av FDA er for det meste risiko, mengden av kontakt med en pasient og deres interne systemer, og om en enhet er avgjørende for å opprettholde livet.,
I tillegg til disse faktorene, FDA vurderer betydelig ekvivalens når du skal bestemme hvordan en enhet er klassifisert. Hvis enheten er lav-risiko og minimalt kontakter pasienten, er du sannsynligvis til å kvalifisere seg for Klasse i og en strømlinjeformet markedet godkjenningsprosessen. Klasse II og III enheter må demonstrere sikkerhet via materielle ekvivalens, et 510(k) innlevering, eller premarket godkjenningsprosessen.
Ved å vite hvordan enheten er klassifisert, du kan effektivisere din vei til markedet godkjenning ved å forstå de prosesser og dokumenter som er sannsynlig å være nødvendig av FDA., Hvis din organisasjon er underlagt en Klasse II og Klasse III 510(k) eller PMA kravet, vil denne kunnskapen kan hjelpe deg med å allokere de riktige ressursene på forhånd og planlegge for en vellykket innlevering.
