Jakie są różnice w klasach urządzeń medycznych FDA?
amerykańska FDA reguluje wszystkie urządzenia medyczne sprzedawane w USA, które są pogrupowane w trzy szerokie klasy. Każdy wyrób medyczny zatwierdzony przez FDA jest klasyfikowany jako klasa I, II Lub III w zależności od ryzyka, inwazyjności i wpływu na ogólny stan zdrowia pacjenta. Ale gdzie są linie narysowane między każdą z tych trzech klas i dlaczego?,
Amerykańskie wytyczne klasyfikacji FDA mogą być bardzo mylące dla producentów urządzeń medycznych z ograniczoną ekspozycją na system. Istnieje ogromna różnica w optymalnej ścieżce do rynku dla producentów w zależności od tego, jak urządzenie jest zgrupowane. Urządzenia klasy i podlegają znacznie mniejszym wymogom regulacyjnym niż urządzenia klasy II Lub III.
rozumiejąc różnice w klasach urządzeń medycznych FDA, możesz zrozumieć, w jaki sposób urządzenie zostanie pogrupowane., Dzięki tej wiedzy producenci wyrobów medycznych na etapie przedsprzedaży mogą lepiej przygotować i przydzielić zasoby potrzebne do zatwierdzenia przez przepisy.
różnice między klasami urządzeń medycznych FDA
FDA sklasyfikowała ponad 1700 różnych typów urządzeń medycznych. Urządzenia są zorganizowane w Kodeksie Przepisów Federalnych (CFR) zgodnie z 16 specjalnościami, takimi jak urządzenia sercowo-naczyniowe lub hematologiczne., Klasyfikacja wyrobu medycznego według jednej z 16 specjalności jest pierwszym krokiem do zrozumienia, czy produkujesz wyrób medyczny klasy I, II Lub III.
Po sklasyfikowaniu urządzenia według specjalności, FDA instruuje producentów, aby przystąpili do powiadomienia przedmarketowego ze wiedzą, czy ich urządzenie jest zwolnione, czy nie. Wyroby medyczne klasy I, kategorii najmniej ryzykowne i inwazyjne, są zwolnione z przedmarketowych procesów powiadamiania. Specjalne urządzenia klasy II są również wyłączone z homologacji przedmarketowej.,
jednak wszystkie urządzenia regulowane przez FDA podlegają aktualnym wymogom Dobrej Praktyki Wytwarzania (cGMP) w zakresie rejestracji, etykietowania i jakości. Ale skąd wiesz, czy Twoje urządzenie jest klasą i czy II i czy musisz przejść przedmarketowe powiadomienie? 
Klasa 1
amerykańska FDA definiuje urządzenia klasy i jako urządzenia, które „nie są przeznaczone do użytku w podtrzymywaniu lub podtrzymywaniu życia lub mają istotne znaczenie w zapobieganiu upośledzeniu zdrowia ludzkiego i nie mogą stanowić potencjalnego nieuzasadnionego ryzyka choroby lub obrażeń.,”
urządzenia te są najczęstszą klasą urządzeń regulowaną przez FDA, stanowiącą 47 procent zatwierdzonych urządzeń na rynku.
urządzenia klasy I mają minimalny kontakt z pacjentami i niewielki wpływ na ogólny stan zdrowia pacjenta. Ogólnie rzecz biorąc, urządzenia klasy I nie wchodzą w kontakt z narządami wewnętrznymi pacjenta, ośrodkowym układem nerwowym lub układem sercowo-naczyniowym. Urządzenia te podlegają najmniejszym wymogom regulacyjnym.,
przykłady urządzeń klasy I:
- Szczoteczka Elektryczna
- depresor języka
- Maska tlenowa
- skalpel chirurgiczny wielokrotnego użytku
- bandaże
- łóżka szpitalne
wprowadzanie urządzeń medycznych klasy i na rynek
urządzenia klasy i są najszybsze i najłatwiejsze do wprowadzenia na rynek, ponieważ stanowią najniższe ryzyko dla pacjenta i rzadko są krytyczne dla opieki podtrzymującej życie. Większość urządzeń klasy i jest zwolniona z wymogów FDA dotyczących powiadamiania Przedmarketowego (510k) i zatwierdzania Przedmarketowego (PMA).,
urządzenia klasy i nie są wyłączone z FDA general controls, serii poleceń, które mają zastosowanie do urządzeń medycznych klasy I, II i III. Przepisy tej ustawy dotyczą zafałszowania, błędnego brandingu, rejestracji urządzeń, zapisów i dobrych praktyk produkcyjnych. Producenci wyrobów medycznych, którzy należą do klasy A, są nadal zobowiązani do wdrożenia systemu zarządzania jakością i przestrzegania standardów w celu zapewnienia jakości produktu.,
podobne czytanie: różnica między powiadomieniem Przedmarketowym 510(k) A zatwierdzeniem Przedmarketowym
wyroby medyczne klasy 2
wyroby medyczne klasy II są bardziej skomplikowane niż wyroby klasy I i stanowią wyższą kategorię ryzyka, ponieważ są bardziej narażone na trwały kontakt z pacjentem. Może to obejmować urządzenia, które wchodzą w kontakt z układu sercowo-naczyniowego pacjenta lub narządów wewnętrznych, i narzędzia diagnostyczne.,
FDA definiuje urządzenia klasy II jako „urządzenia, dla których ogólne kontrole są niewystarczające, aby zapewnić wystarczającą pewność bezpieczeństwa i skuteczności urządzenia.,zestawy est
wprowadzanie na rynek urządzeń medycznych klasy II
kontrole mogą się różnić w zależności od urządzenia, ale według FDA mogą obejmować:
- wydajność urządzenia
- Nadzór po sprzedaży
- Kontrola pacjenta rejestry
- specjalne wymagania dotyczące etykietowania
- wymagania dotyczące danych przedmarketowych
- wytyczne
większość urządzeń klasy II jest zatwierdzona przez FDA na rynek poprzez powiadomienie przedmarket lub proces 510(K).,
urządzenia klasy II podlegają tym samym ogólnym kontrolom, o których mowa powyżej, ale FDA definiuje je jako ” urządzenia, dla których ogólne kontrole są niewystarczające, aby zapewnić wystarczającą pewność bezpieczeństwa i skuteczności urządzenia.”Z tego powodu urządzenia klasy II podlegają również specjalnym kontrolom. Przepisy te zależą od urządzenia i mogą obejmować specjalne wymagania dotyczące etykietowania, rejestry pacjentów i standardy wydajności.
większość urządzeń klasy II wchodzi na rynek za pomocą procesu powiadamiania Przedmarketowego (510k)., 510 (k) jest złożoną aplikacją do FDA, która pokazuje, że urządzenie jest bezpieczne i skuteczne, wykazując, że urządzenie jest równoważne z innym urządzeniem, które jest na rynku.
proces ten polega na wykazaniu „istotnej równoważności” z innym urządzeniem, które jest znane w FDA jako „predykat.”Nie oznacza to, że urządzenia muszą być identyczne, ale wymagają znaczących podobieństw w użyciu, projekcie, materiałach,etykietowaniu, standardach i innych cechach.,
FDA opublikowała listę zwolnień na początku 2018 roku, która wyłącza ponad 800 generycznych wyrobów medycznych klasy I I II z procesu 510(k). Jeśli masz generyczny wyrób medyczny klasy II, możesz dowiedzieć się, czy jest on zwolniony z zgłoszenia 510(k), przeszukując bazę danych klasyfikacji produktów FDA.
podobne czytanie: 5 powodów, dla których FDA 510(k) to świetny ruch.
Klasa 3
FDA definiuje urządzenia klasy III jako produkty, które „zazwyczaj podtrzymują lub podtrzymują życie, są wszczepiane lub stwarzają potencjalne nieuzasadnione ryzyko choroby lub urazu.,”
tylko 10 procent urządzeń regulowanych przez Amerykańską FDA zalicza się do klasy III. klasyfikacja ta jest na ogół rozszerzona na trwałe implanty, inteligentne urządzenia medyczne i systemy podtrzymywania życia.
chociaż Klasa III jest zazwyczaj zarezerwowana dla najbardziej innowacyjnych i najnowocześniejszych urządzeń medycznych, istnieją inne urządzenia, które mogą z różnych powodów zaliczyć do klasy III., Niektóre urządzenia, które są początkowo klasyfikowane jako klasa II, mogą zostać podniesione do klasy III, jeśli producent nie jest w stanie wykazać istotnej równoważności z predykatem (istniejącym produktem) podczas procesu składania PMA (510k).,
przykłady wyrobów medycznych klasy III:
- implanty piersi
- stymulatory serca
- defibrylatory
- respiratory wysokiej częstotliwości
- implanty ślimakowe
- monitory pobierania próbek krwi płodu
- implantowana protetyka
wprowadzenie na rynek wyrobów medycznych klasy III
urządzenia klasy III są przedmiotem do wszystkich kontroli ogólnych FDA i procesu zatwierdzania przedmarketowego (PMA) FDA., FDA pisze: „ze względu na poziom ryzyka związanego z urządzeniami klasy III, FDA ustaliła, że ogólne i specjalne kontrole same są niewystarczające, aby zapewnić bezpieczeństwo i skuteczność urządzeń klasy III.”
PMA jest najbardziej intensywnym rodzajem aplikacji marketingowej wymaganej przez FDA. Niektóre urządzenia klasy III FDA są zwolnione i mogą kwalifikować się do zgłoszenia 510(k), ale oczekuje się, że większość uzyska aprobatę Przedmarketową.,
proces PMA wymaga rygorystycznego badania wyrobu medycznego w celu udowodnienia bezpieczeństwa i skuteczności poprzez opracowanie opartego na danych profilu korzyści / ryzyka. Proces PMA zazwyczaj obejmuje badania kliniczne oraz znaczny czas i zasoby na wystarczające zbieranie danych. Jedynymi wyjątkami od procesu PMA w ramach klasy III są urządzenia o znaczącym odpowiedniku. Możesz określić, czy urządzenie klasy III może być sprzedawane z 510(k), przeszukując bazę danych FDA Przedmarket Approval (PMA) i bazę danych 510(k) Przedmarket Notification.,
Jak określić swoją klasę
pierwszym krokiem w kierunku klasyfikacji wyrobu medycznego jest zapoznanie się z przepisami klasyfikacji FDA, listą 16 kategorii wyrobów medycznych według specjalizacji medycznej.
jako przykład pokażemy Ci kroki w celu określenia klasyfikacji alarmu ciśnienia krwi. Urządzenie jest sklasyfikowane w kategorii 870: urządzenia sercowo-naczyniowe.,
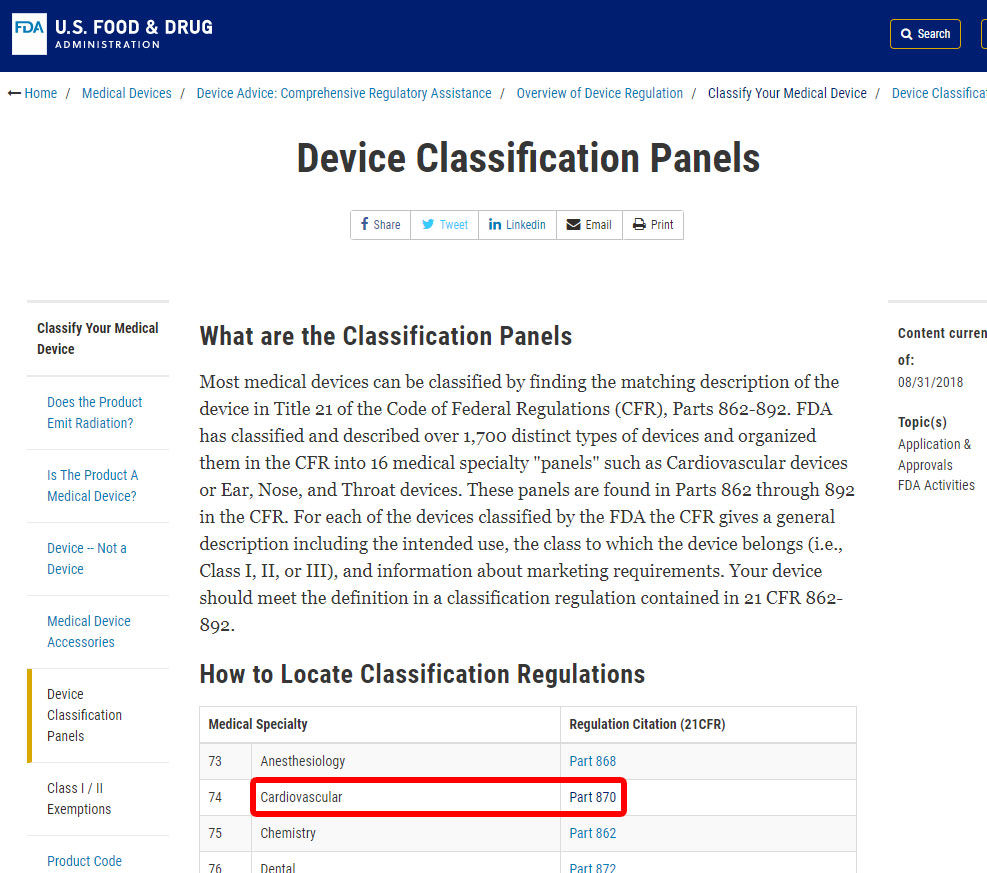
po zlokalizowaniu odpowiedniej specjalności medycznej Kliknij kategorię i przejdź do listy urządzeń, aż znajdziesz odpowiedni kod urządzenia i powiązany z nim kod.
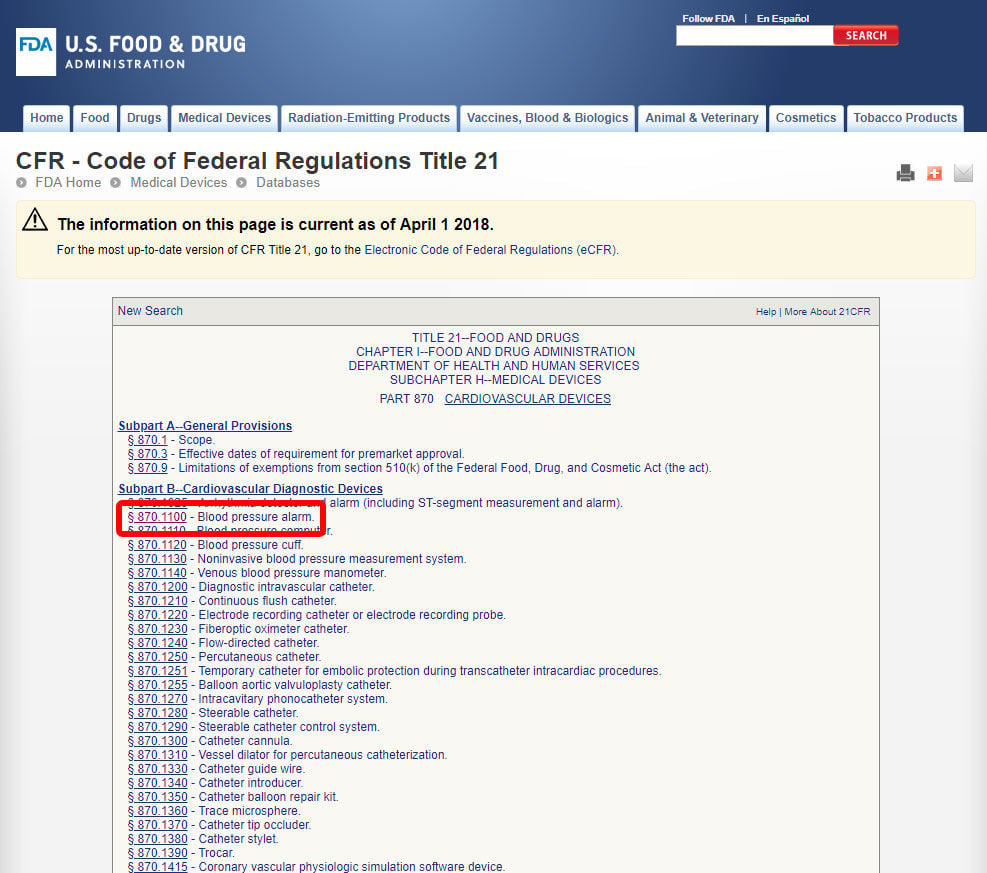
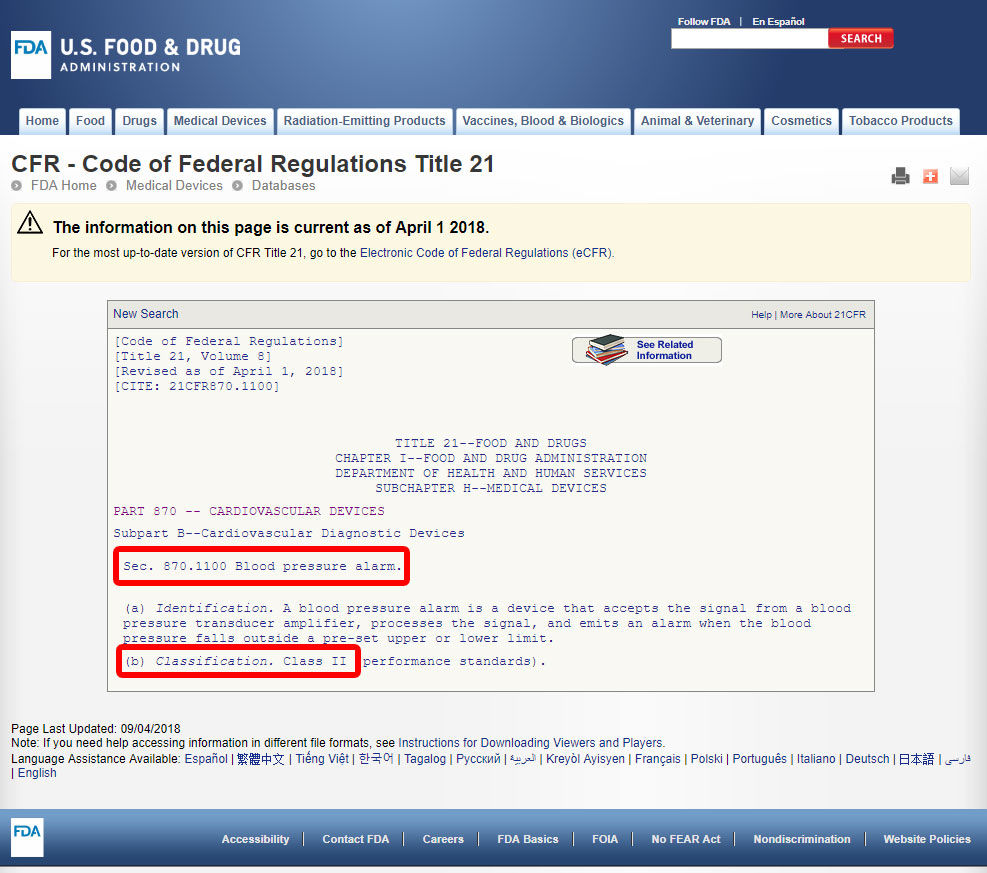
kliknij kod urządzenia i otwórz wytyczne. Klasyfikacja wyrobów jest wymieniona w sekcji (b).,
Jeśli Twoje urządzenie nie ma na liście odpowiednika wśród 1700 urządzeń sklasyfikowanych przez FDA, najprawdopodobniej jest to innowacyjne urządzenie bez znaczącego odpowiednika i zostałoby sklasyfikowane jako klasa III.
zrozumienie klas urządzeń medycznych FDA
różnice między urządzeniami medycznymi sklasyfikowanymi przez FDA jako klasa I, II lub III są głównie związane z ryzykiem, ilością kontaktu z z pacjentem i jego systemami wewnętrznymi oraz czy urządzenie ma kluczowe znaczenie dla podtrzymania życia.,
oprócz tych czynników, FDA bierze pod uwagę istotną równoważność przy określaniu sposobu klasyfikacji urządzenia. Jeśli Twoje urządzenie jest niskiego ryzyka i w minimalnym stopniu kontaktuje się z pacjentem, prawdopodobnie kwalifikujesz się do klasy I i usprawnionego procesu zatwierdzania rynku. Urządzenia klasy II i III muszą wykazywać bezpieczeństwo poprzez równoważność merytoryczną, zgłoszenie a 510 (k) lub przedmarketowy proces zatwierdzania.
wiedząc, w jaki sposób urządzenie jest klasyfikowane, możesz usprawnić swoją drogę do zatwierdzenia rynkowego poprzez zrozumienie procesów i dokumentów, które mogą być wymagane przez FDA., Jeśli Twoja organizacja podlega wymogowi klasy II lub klasy III 510(k) lub PMA, wiedza ta może pomóc ci wcześniej przydzielić odpowiednie zasoby i zaplanować udane zgłoszenie.
