Holoprosencefalia e Strabismus
Pavlina S. Kemp, MD, Grant Casey, Susannah Q. Longmuir, MD
12 de junho de 2012
queixa-chefe: cruzamento ocular
história da doença actual
O doente é uma mulher de 15 meses de idade à apresentação na clínica ocular, com história de hidrocefalia grave ao nascimento. Ela também foi diagnosticada com holoprosencefalia alobar no nascimento com convulsões. Ela foi originalmente indicada para cruzar os olhos. Apresentamos – lhe um curso clínico notável.,
História Médica:
- Hidrocefalia s/p derivação ventriculoperitoneal
- Holoprosencefalia (alobar tipo)
- desordem de Apreensão
Passado História Cirúrgica:
- colocação de derivação Ventriculoperitoneal, 2004
- shunt Ventriculoperitoneal revisão, 1/2005
- shunt Ventriculoperitoneal revisão, 6/2005
a História da Família: Não conhecida a história da família de holoprosencefalia, a ambliopia e estrabismo. História Social: o doente vive em casa com os pais e duas irmãs.,
Medicamentos: None
Exame e Curso Clínico:
Idade: 15 meses
a Acuidade Visual Central, instável e mantido OD e central, instável e mantidos OS
Caixa de acuidade de teste:
- Sem correção UO: 20/800
Alunos: Igualmente rodada e rapidamente reativa, não em relação defeito pupilar aferente.,
de Visão Estéreo: não é Possível teste
Mobilidade e Estrabismo:
- Grande variável de esotropia
- Bilaterais de elevação e abdução de déficits
- Intermitente horizontal nistagmo
Cycloplegic Refração:
- OD: +4.00
- OS: +6.00
Externo Exame: Notável pelo grande perímetro cefálico
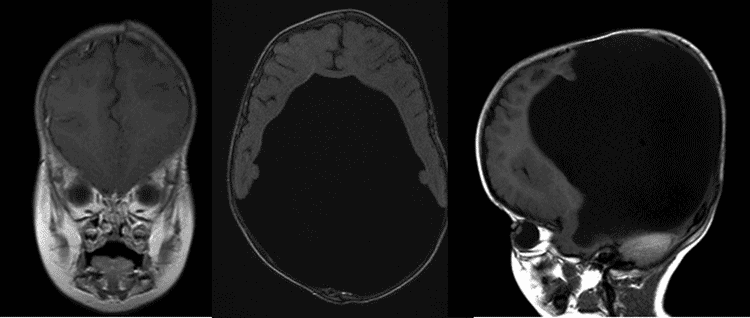
Lâmpada de Fenda Exame: Normal segmento anterior do exame ou sem evidência de catarata ou outras mídias, opacidades. nervos ópticos com aparência normal e exame do fundo dilatado normal. Não há sinais de hipoplasia do nervo óptico em nenhum dos olhos., Figura 1: IRM Axial e saggital ilustrando o aumento dos ventrículos secundários à hidrocefalia.
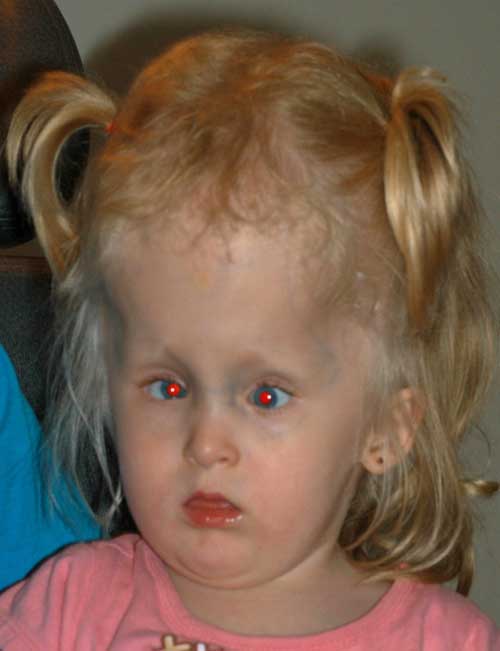
este ponto, após discussão com sua família, a cirurgia para estrabismo foi adiada e a correção de sua hiperopia foi tentada. Os óculos foram prescritos. Ela não era capaz de usar óculos confortavelmente e lentes de contato foram tentadas. O Patching foi realizado para tratar a ambliopia. A paciente foi seguida e aos 3 anos, seus pais queriam prosseguir com a cirurgia do estrabismo para “descruzar” seus olhos.,
Idade: 3
a Acuidade Visual Central, CUSM OD e CUSM OS
Mobilidade e Estrabismo:
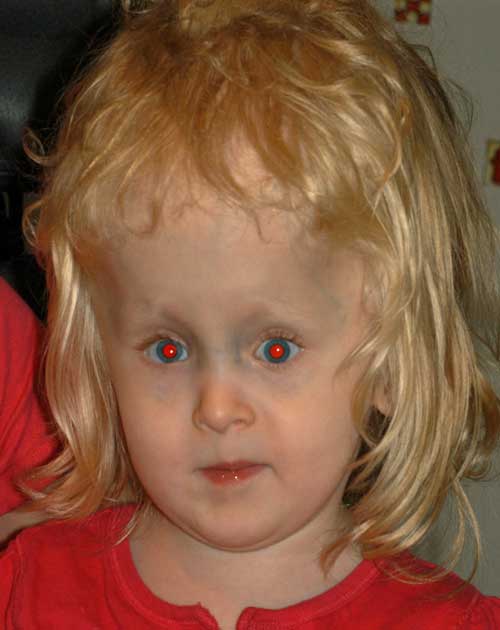
- Grande ângulo de esotropia de 55-60 prisma dioptrias (Figura 2)
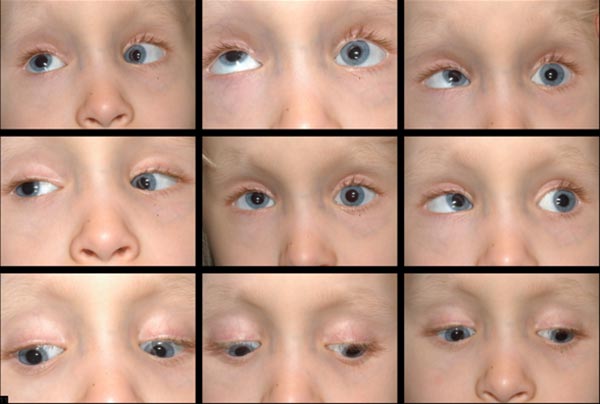
- Bilaterais rapto déficits e uma esquerda monocular elevação de deficiência
no momento da cirurgia do estrabismo, as ducções forçadas intra-operatórias mostraram restrições tanto dos músculos do recto medial e o paciente teve uma inserção anômala do músculo do recto medial. A inserção muscular do rectus medial antes da deserção foi encontrada a 7 mm do limbus (mais posterior do que o esperado 5, 5 mm do limbus). Secundária à anatomia não ser normal, uma abordagem conservadora foi realizada e ela passou por rectos mediais bilaterais de 5,5 mm, deixando o recto medial aos 12 anos.,A 5 mm do limbus. no pós-operatório, teve uma pequena esotropia residual com pequeno desvio vertical e uma deficiência elevatória mais visível do olho esquerdo.
idade 6:
seus pais estavam felizes com o alinhamento; no entanto, com o tempo, ela desenvolveu uma notável hipertropia direita. Aos 6 anos de idade, sua família decidiu prosseguir com uma segunda cirurgia estrabismo para resolver o desalinhamento vertical.,
uma recessão recto inferior esquerda vs uma recessão recto superior direito foi planejada, com decisão sobre qual procedimento seria realizado com base em ducções forçadas intra-operatórias. As condutas forçadas Intra-operatórias mostraram um rectus inferior esquerdo apertado, o que é consistente com a deficiência de elevação monocular que tínhamos considerado anteriormente. Um recto inferior esquerdo de 6 mm foi realizado, movendo o músculo de 8 mm posterior ao limbo, onde foi encontrado, para 14 mm posterior ao limbo.,
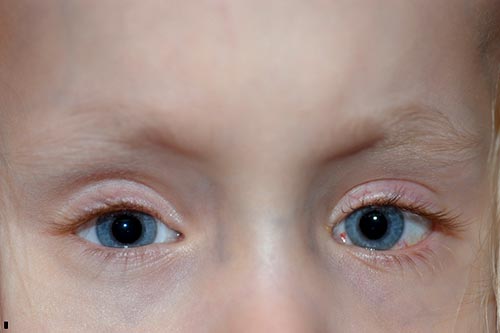
lguns meses após sua segunda cirurgia, sua mãe começou a notar que seu olho direito flutuou para cima em tempos de desatenção. Ela foi vista na clínica e notou um manifesto Desvio vertical dissociado à direita., Decidiu-se prosseguir novamente com a cirurgia do estrabismo e uma recessão recto superior direita de 8 mm pelo método do hangback, movendo o músculo de 8 mm posterior ao limbo, onde foi encontrado, para 16 mm posterior ao limbo.
o paciente saiu-se bem no pós-operatório (Figura 6), e tem vindo a desenvolver progressivamente as suas capacidades visuais. Ela tem feito notavelmente bem em geral e está andando e agora lendo.
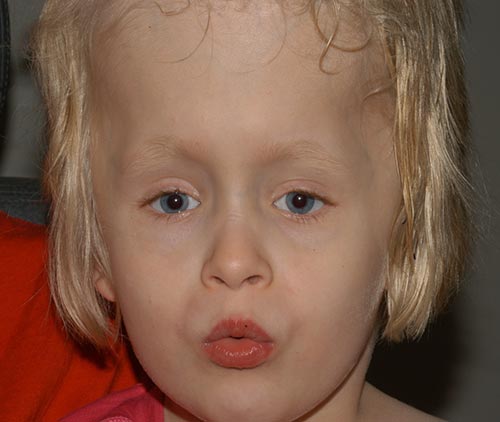
diagnóstico:
Holoprosencefalia com esotropia infantil e Estrabismo complexo, com variabilidade nas inserções musculares do recto medial.
discussão:
Holoprosencefalia é um tipo de transtorno cefálico caracterizado pela falha no desenvolvimento do prosencefalo embrionário, levando a uma estrutura cerebral de um só umbigo e graves defeitos no crânio e na face., Existem três classificações de holoprosencefalia: holoprosencefalia alobar, holoprosencefalia semi-lobar e holoprosencefalia lobar. a holoprosencefalia Alobar é responsável por dois terços dos pacientes afetados, e é a forma mais grave, caracterizada pela falha do cérebro em se separar em duas metades. Isto resulta em um único ventrículo primitivo, ausência de bolbos olfativos e tractos ópticos e anomalias graves no desenvolvimento. É geralmente associado com graves anomalias faciais, incluindo olhos bem espaçados, tamanho pequeno da cabeça, fenda labial e palato., A holoprosencefalia Semi-lobar, responsável por um quarto dos casos de holoprosencefalia, é uma forma intermediária da doença e é caracterizada por hemisférios cerebrais parcialmente separados e um único ventrículo. A holoprosencefalia Lobar é a forma menos grave, na qual o cérebro do paciente pode ser quase normal; há uma fissura distinta entre os lobos centrais desenvolvidos, e alguma fusão das estruturas cerebrais está presente (Nanni, 2000). Na maioria dos casos de holoprosencefalia, as malformações cerebrais são incompatíveis com a vida., Em casos menos graves, os bebês nascem com desenvolvimento cerebral normal ou quase normal e vários graus de deformidade facial. os defeitos craniofaciais da linha média são a marca da holoprosencefalia e podem incluir microcefalia, hipotelorismo (olhos anormalmente espaçados), anomalias nasais, tais como achatamento nasal ou um único naris, e os defeitos do lábio superior e palato, tais como fenda palatina ou um único incisivo frontal. A ciclopia pode estar presente nas formas mais severas, onde um probóscide semelhante ao nariz está presente sobre um único olho no meio da face (Nanni, 2000)., Pensa-se que o grau de deformidade facial indica a gravidade dos defeitos intracranianos. Co-morbilidades associadas incluem disfunção da glândula pituitária e hipotálamo, resultando em desregulação da temperatura corporal, convulsões e atraso mental de gravidade variável (Dubourg, 2007). Também foram observados hipotonia e distonia (Barkovich, 2002).a Holoprosencefalia ocorre durante as primeiras semanas de vida intra-uterina. A prevalência da holoprosencefalia no desenvolvimento embrionário precoce é de 1:250, diminuindo para 1:10,000-1:20,000 no termo (Nanni, 2000)., Não há uma causa conhecida de holoprosencefalia, embora tenha havido muitas propostas fatores de risco, incluindo diabetes materno (1% de risco, 200 vezes de aumento) (Barra, 1983), infecções durante a gravidez, como a TOCHA infecções (Munke, 1989), e a exposição a substâncias tóxicas, incluindo o álcool, lítio, Thorazine, hormônios, anticonvulsivantes e retinoic (Nanni, 2000). A maioria dos casos são considerados esporadicamente, embora a holoprosencefalia também tenha uma base genética., A holoprosencefalia familiar tem sido vista herdada em ambos os padrões autossômicos dominantes e autossômicos recessivos. Anomalias cromossômicas também têm sido associadas com a holoprosencefalia, sendo a trissomia 13 a mais comum, embora esta não seja uma associação constante (Kallen, 1992).acredita-se que a patogênese envolva um defeito nos genes sinalizadores responsáveis pela regulação do padrão do tubo neural., Os achados intracranianos incluem hipoplasia cortical variando, fusão do diencéfalo, gânglios basais e tálamo e presença de um quisto dorsal (resultante de talami fundido) expandindo-se a partir de um terceiro ventrículo parcialmente bloqueado (Simon, 2001). A hidrocefalia, causada por acumulação anormal de líquido cefalorraquidiano nos ventrículos, não é incomum na holoprosencefalia e pensa-se ser devido a malformação dos ventrículos ou produção excessiva de líquido cefalorraquidiano., Isso muitas vezes complica a classificação de holoprosencefalia, como o cérebro é comprimido e o crânio anteriormente microcéfalo é permitido expandir antes da fusão das suturas cranianas (Tripathi, 2009). É importante abordar a visão do doente para permitir uma interacção óptima com o seu ambiente circundante. Muitas vezes os óculos não são tolerados devido ao grau de assimetria facial e anormalidades estruturais presentes. Nestas situações, achamos que as lentes de contato devem ser consideradas como uma forma de melhorar a função visual.,
embora haja variação da inserção muscular do recto medial na população em geral, este caso é notável para as inserções musculares extraoculares anômalas, especificamente a distância de 7 mm do reti medial a partir do limbo. Como mencionado acima, defeitos da linha média são comuns na holoprosencefalia, o que pode explicar por que o reti medial foi preferencialmente envolvido. Com exceção da ciclopia, pouco foi publicado sobre associações oculares e estrabismicas com a holoprosencefalia.,globalmente, o tratamento é altamente individualizado com base na gravidade e configuração da malformação do doente. O tratamento é de suporte e sintomático e o prognóstico depende muito do tipo de holoprosencefalia e suas anomalias associadas (Nanni, 2000).no caso apresentado acima, uma das considerações mais significativas para a família do paciente foi como intervir para melhorar a visão e permitir o crescimento e desenvolvimento contínuos. O paciente estava gravemente doente nos primeiros anos de vida, e os cuidados paliativos foram inicialmente discutidos como uma opção viável., A família do paciente queria continuar o tratamento e procurar possíveis intervenções para melhorar a qualidade de vida. Quando confrontados com situações difíceis como esta, é essencial que os prestadores de cuidados de saúde ajudem as famílias a tomar decisões num ambiente respeitoso sem julgamento ou influência indevida. The American College of Critical Care Medicine Task Force published clinical practice guidelines addressing support of the family in the patient-centered intensive care unit (Davidson, 2007)., Através de uma boa comunicação, gestão de conflitos e habilidades de facilitação de reuniões, as famílias podem estar envolvidas em um modelo de tomada de decisão compartilhada, no qual as famílias não são as únicas responsáveis por todas as decisões médicas autonomamente, nem os prestadores de cuidados paternalistas. Durante as reuniões familiares, recomenda-se que os membros da família sejam questionados sobre a sua compreensão dos cuidados do paciente, dos seus medos e das estratégias de enfrentamento., Os prestadores de cuidados de saúde são encorajados a repetir os sentimentos da família para permitir o desenvolvimento da confiança na equipa e no processo de tomada de decisão. Depois disso, os profissionais devem fornecer informações claras e honestas em linguagem acessível, com a oportunidade de fazer perguntas. O objetivo da discussão é o consenso, que é ajudado pelo reconhecimento respeitoso de todas as opiniões.,
Penticuff e Arheart estudou a eficácia das reuniões entre prestadores de cuidados de saúde e os pais em uma unidade de cuidados intensivos neonatais definição, e mostrou que a tomada de decisões compartilhada resultou em menos conflitos, irreais expectativas de pais e familiares, e melhor colaboração, bem como ajudar os pais a compreender melhor seu filho, a situação médica (Penticuff, 2005). Os níveis de estresse familiar são mostrados ser reduzidos com comunicação aberta e eficaz, bem como um ambiente de esperança (Davidson, 2007)., O que é importante é que esses cuidados centrados em pacientes têm sido mostrados para melhorar os resultados clínicos, bem como (Lewin, 2001).diagnóstico diferencial deficiência de elevação Monocular Desvio vertical dissociado
resumo
- Holoprosencefalia é um grupo de perturbações resultantes de uma anomalia no desenvolvimento embrionário do cérebro.a Holoprosencefalia está associada a anomalias cranianas e faciais, incluindo o estrabismo.,possivelmente, a variabilidade na anatomia muscular intra-ocular pode estar relacionada com as anomalias no desenvolvimento embrionário.
Sinais
- anomalias Faciais incluem espaçados olhos, o pequeno tamanho da cabeça, de lábio e palato
- a Hidrocefalia pode levar ao grande tamanho da cabeça
- Anômala inserções de extraocular músculos pode levar ao estrabismo
Sintomas
- Geralmente observado ao nascimento
- Incluir a temperatura do corpo desregulação, convulsões, retardo mental, de gravidade variável, hipotonia e distonia.,
Tratamento
- Individualizada para cada paciente de caso
- shunt Ventriculoperitoneal para hidrocefalia
- o Estrabismo de cirurgia, se indicado
- lentes de Contato para correção de refração
Barkovich AJ, Simon EM, Clegg NJ, Parente SL, Hahn JS. Análise do córtex cerebral na holoprosencefalia com atenção às fissuras sylvianas. AJNR Am J Neuroradiol. 2002;23(1):143-50.Blaas HG, Eriksson AG, Salvesen KA, Isaksen CV, Christensen B, Møllerløkken G, Eik-Nes SH., Cérebros e rostos em holoprosencefalia: descrição pré e pós – natal de 30 casos. Ecografia Obstetrícia Ginecol. 2002;19(1):24-38.Barra Jr M, Hanson JW, Currey K, Sharp S, Toriello H, Schmickel RD, Wilson GA. Holoprosencefalia em crianças de mães diabéticas. J Pediatr 1983; 102: 565D8.Davidson JE, Powers K, Hedayat KM, Tieszen M, Kon AA, Shepard e, Spuhler V, Todres ID, Levy M, Barr J, Ghandi R, Hirsch G, Armstrong D., Clinical practice guidelines for support of the family in the patient-centered intensive care unit: American College of Critical Care Medicine Task Force 2004-2005. American College of Critical Care Medicine Task Force 2004-2005, Society of Critical Care Medicine. Hematócrito Med. 2007;35(2):605-22.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencefalia. Orphanet J Rare Dis. 2007 2;2:8.Kallen B, Castilla EE, Lancaster PAL, et al. The cyclops and the mermaid: an epidemiological study of two types of rare malformações. J Med Genet 1992; 29: 30-35.,
Lewin SA, Skea ZC, Entwistle V, Zwarenstein M, Dick J. Interventions for providers to promote a patient-centred approach in clinical consultations. Cochrane Database Syst Rev. 2001; (4): CD003267. (PMID:11687181)
Munke M. Clinical, citogenetic and molecular approaches to the genetic heterogeneity of holoprosencephalia. Am J Med Genet 1989; 34: 237-245.Nanni L, Schelper RL, Muenke MT. Genética Molecular da holoprosencefalia. Front Biosci. 2000 1; 5: D334-42.Penticuff JH, Arheart Kl.,Eficácia de uma intervenção para melhorar a colaboração entre pais e profissionais em cuidados intensivos neonatais. J Perinat Nurs Neonatal. 2005;19(2):187-202.Scott WE, Jackson OB. Paralisia do elevador duplo: o significado da restrição do rectus inferior. Am Orthopt J. 1977; 27: 5-10.Simon em, Hevner RF, Pinter J, Clegg NJ, Delgado M, Kinsman SL, Hahn JS, Barkovich AJ. O quisto dorsal na holoprosencefalia e o papel do tálamo na sua formação. Neuroradiologia. 2001;43(9):787-91.Tripathi AK, Agrawal D, Sedain G. holoprosencefalia hidrocefálica: um oximoro?, Insights into etiology and management. Neurociência Pediatra. 2009;4(1):41-3.consentimento para a utilização de fotografias e vídeos obtidos da mãe do doente.
sugerido formato de citação: Kemp PS, Casey, G, Longmuir SQ. Holoprosencefalia e Strabismus.Eyerounds.org. June 12, 2012, Available from: http://EyeRounds.org/cases/151-holoprosencephaly-strabismus.htm