Quais são as diferenças nas aulas de dispositivos médicos da FDA?
the US FDA regulates all medical devices marketed in the US, which are grouped into three broad classes. Qualquer dispositivo médico aprovado pela FDA é classificado como Classe I, II ou III, dependendo do risco do dispositivo, inviabilidade e impacto na saúde geral do paciente. Mas onde estão as linhas traçadas entre cada uma destas três classes, e por quê?,
as Diretrizes de classificação da FDA dos EUA podem ser altamente confusas para os fabricantes de dispositivos médicos com exposição limitada ao sistema. Há uma enorme diferença no caminho ideal para o mercado para os fabricantes, dependendo de como o seu dispositivo é agrupado. Os dispositivos da classe I estão sujeitos a requisitos regulamentares muito menos rigorosos do que os dispositivos das Classes II ou III.
ao compreender as diferenças nas classes de dispositivos médicos da FDA, você pode entender como o seu dispositivo será agrupado., Com este conhecimento em mãos, os fabricantes de dispositivos médicos nas fases de pré-comercialização podem preparar e alocar melhor os recursos necessários para a aprovação regulamentar.
diferenças entre Classes de dispositivos médicos da FDA
a FDA classificou mais de 1.700 tipos distintos de dispositivos médicos. Os dispositivos estão organizados no código de Regulamentos Federais (CFR) de acordo com 16 especialidades, tais como dispositivos cardiovasculares ou hematológicos., Classificar o seu dispositivo médico de acordo com uma das 16 especialidades é o primeiro passo para compreender se está a fabricar um dispositivo médico de classe I, II ou III.
Depois de classificar um dispositivo de acordo com a especialidade, a FDA instrui os fabricantes a proceder à notificação pré-comercialização com conhecimento de se seu dispositivo está isento ou não. Os dispositivos médicos de classe i, a categoria menos arriscada e invasiva, estão isentos de processos de notificação pré-comercialização. Os dispositivos específicos da classe II estão igualmente isentos da aprovação antes da comercialização.,
no entanto, todos os dispositivos regulados pela FDA estão sujeitos aos atuais requisitos de Boas Práticas de fabricação (cGMP) para registro, rotulagem e qualidade. Mas como é que sabe se o seu dispositivo é de classe I ou II, e se é obrigado a submeter-se a notificação antes do mercado? 
Classe 1
a FDA dos EUA define dispositivos de classe I como dispositivos que “não se destinam a ser utilizados no suporte ou sustentação de vida ou de importância substancial na prevenção de danos para a saúde humana, e eles podem não apresentar um potencial risco irrazoável de doença ou lesão.,”
estes dispositivos são a classe mais comum de dispositivos regulados pela FDA, constituindo 47 por cento dos dispositivos aprovados no mercado.os dispositivos da classe I têm um contacto mínimo com os doentes e um baixo impacto na saúde geral do doente. Em geral, os dispositivos de classe I não entram em contacto com os órgãos internos do doente, o sistema nervoso central ou o sistema cardiovascular. Estes dispositivos estão sujeitos aos menores requisitos regulamentares.,
Exemplos de Dispositivos Classe I:
- Escova de dentes Elétrica
- Língua Opióide
- Máscara de Oxigênio
- Reutilizáveis Bisturi Cirúrgico
- Curativos
- Camas de Hospital
Trazendo Classe I Dispositivos Médicos para o Mercado
Dispositivos classe I é o mais rápido e mais fácil de trazer para o mercado uma vez que eles apresentam a menor quantidade de risco para o paciente e raramente são essenciais para manter a vida de cuidados. A maioria dos dispositivos de classe I estão isentos dos requisitos da FDA para notificação pré-comercialização (510k) e aprovação pré-comercialização (PMA).,
os dispositivos da classe I não estão isentos dos controlos gerais da FDA, uma série de comandos que se aplicam aos dispositivos médicos das classes I, II e III. As disposições da presente lei abordam a adulteração, a má marca, o registo dos dispositivos, os registos e as boas práticas de fabrico. Os fabricantes de dispositivos médicos que entram na classe A ainda são obrigados a implementar um sistema de gestão da qualidade e seguir os padrões para garantir um produto de qualidade.,
LER RELACIONADO: A Diferença Entre Premarket Notificação de 510(k) e Premarket Aprovação
de Classe 2
dispositivos médicos da Classe II são mais complicados do que dispositivos Classe I e apresentam uma maior categoria de risco, porque eles são mais propensos a entrar em contato prolongado com um doente. Isto pode incluir dispositivos que entram em contato com o sistema cardiovascular do paciente ou órgãos internos, e ferramentas de diagnóstico.,
A FDA define dispositivos de classe II como ” dispositivos para os quais os controlos gerais são insuficientes para proporcionar uma garantia razoável da segurança e eficácia do dispositivo.,est Kits
Trazendo Dispositivos Médicos da Classe II para o Mercado
Controles variam consoante o dispositivo, mas de acordo com o FDA, podem incluir:
- desempenho do Dispositivo
- Postmarket de vigilância
- registos de doentes
- Especiais requisitos de rotulagem
- Premarket requisitos de dados
- Orientações
A maioria dos dispositivos de Classe II são aprovados pela FDA para o mercado, por meio da Premarket Notificação, ou 510(k) do processo.,
os dispositivos de classe II estão sujeitos aos mesmos controlos gerais acima mencionados, mas a FDA define-os como sendo “dispositivos para os quais os controlos gerais são insuficientes para proporcionar uma garantia razoável da segurança e eficácia do dispositivo.”Por essa razão, os dispositivos da classe II também estão sujeitos a controlos especiais. Estes regulamentos dependem do dispositivo e podem incluir requisitos especiais de rotulagem, registros de pacientes e padrões de desempenho.
a maioria dos dispositivos da classe II vêm ao mercado usando o processo de notificação pré-mercado (510k)., A 510(k) é uma aplicação complexa para o FDA, o que demonstra que um dispositivo é seguro e eficaz, demonstrando que o dispositivo é equivalente a outro dispositivo, que está no mercado.
Este processo envolve mostrar ” equivalência substancial “a outro dispositivo que é conhecido na linguagem FDA como “o predicado”.”Isso não significa que os dispositivos precisam ser idênticos, mas eles exigem semelhanças significativas no uso, design, materiais, rotulagem, padrões e outras características.,
A FDA publicou uma lista de isenção no início de 2018, que isenta mais de 800 dispositivos médicos genéricos das classes I e II do processo 510(k). Se você tem um dispositivo médico genérico de classe II, você pode descobrir se ele está isento de um arquivo 510(k), pesquisando a base de dados de classificação de produtos da FDA.
reading RELATED: 5 Reasons Overhauling FDA 510 (k) is a Great Move.
Classe 3
a FDA define dispositivos de classe III como produtos que ” normalmente sustentam ou suportam a vida, são implantados ou apresentam um potencial risco irrazoável de doença ou lesão.,”
apenas 10% dos dispositivos regulados pela FDA dos EUA caem na classe III. esta classificação é geralmente estendida aos implantes permanentes, dispositivos médicos inteligentes e sistemas de suporte de vida. embora a classe III esteja geralmente reservada para os dispositivos médicos mais inovadores e de vanguarda, existem outros dispositivos que podem ser classificados na classe III por diferentes razões., Alguns dispositivos que são classificados inicialmente como Classe II podem ser aumentados para Classe III se o fabricante não puder demonstrar equivalência substancial a um predicado (produto existente) durante o processo de depósito PMA (510k).,
Exemplos de Classe III, de Dispositivos Médicos:
- implantes de Mama
- marca-passos
- Desfibriladores
- Alta-frequência ventiladores
- implante Coclear
- sangue Fetal amostragem monitores
- Implantados próteses
Trazendo Classe III de Dispositivos Médicos para o Mercado
Classe III dispositivos estão sujeitos a todas as FDA Controles Gerais e o FDA Premarket Aprovação (PMA) do processo., A FDA escreve: “devido ao nível de risco associado aos dispositivos de classe III, A FDA determinou que os controles gerais e especiais por si só são insuficientes para garantir a segurança e eficácia dos dispositivos de classe III.”
O PMA é o tipo mais intensivo de aplicação de marketing de dispositivo requerido pela FDA. Alguns dispositivos da classe III da FDA estão isentos e podem se qualificar para um arquivo 510(k), mas a maioria é esperada para ganhar a aprovação do pré-mercado.,o processo PMA requer um estudo rigoroso de um dispositivo médico para provar a segurança e a eficácia através do desenvolvimento de um perfil de benefício/risco baseado em dados. O processo PMA geralmente envolve ensaios clínicos e tempo e recursos significativos para uma recolha de dados suficiente. As únicas exceções ao processo PMA dentro da classe III são dispositivos com um equivalente substancial. Você pode determinar se um dispositivo de classe III pode ser comercializado com um 510(k), pesquisando a base de dados de aprovação de Premarket (PMA) da FDA e a base de dados de notificação de Premarket 510(k).,
como determinar a sua classe
o primeiro passo para classificar o seu dispositivo médico é navegar pelos Regulamentos de classificação da FDA, a lista de 16 categorias para dispositivos médicos de acordo com a especialização médica.como exemplo, vamos mostrar-lhe os passos para identificar a classificação de um alarme de pressão arterial. O dispositivo está classificado na categoria 870: dispositivos cardiovasculares.,
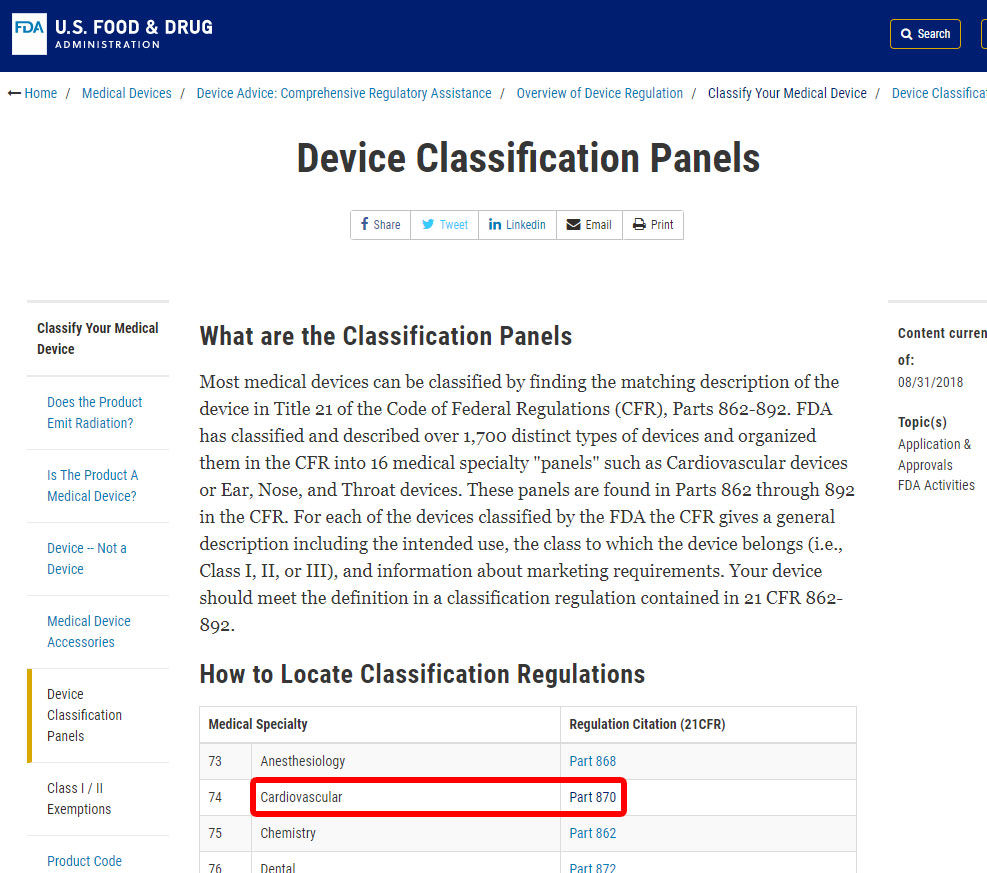
Depois de ter localizado a especialidade médica relevante, clique na categoria e navegue na lista de dispositivos até encontrar um equivalente e o código associado.
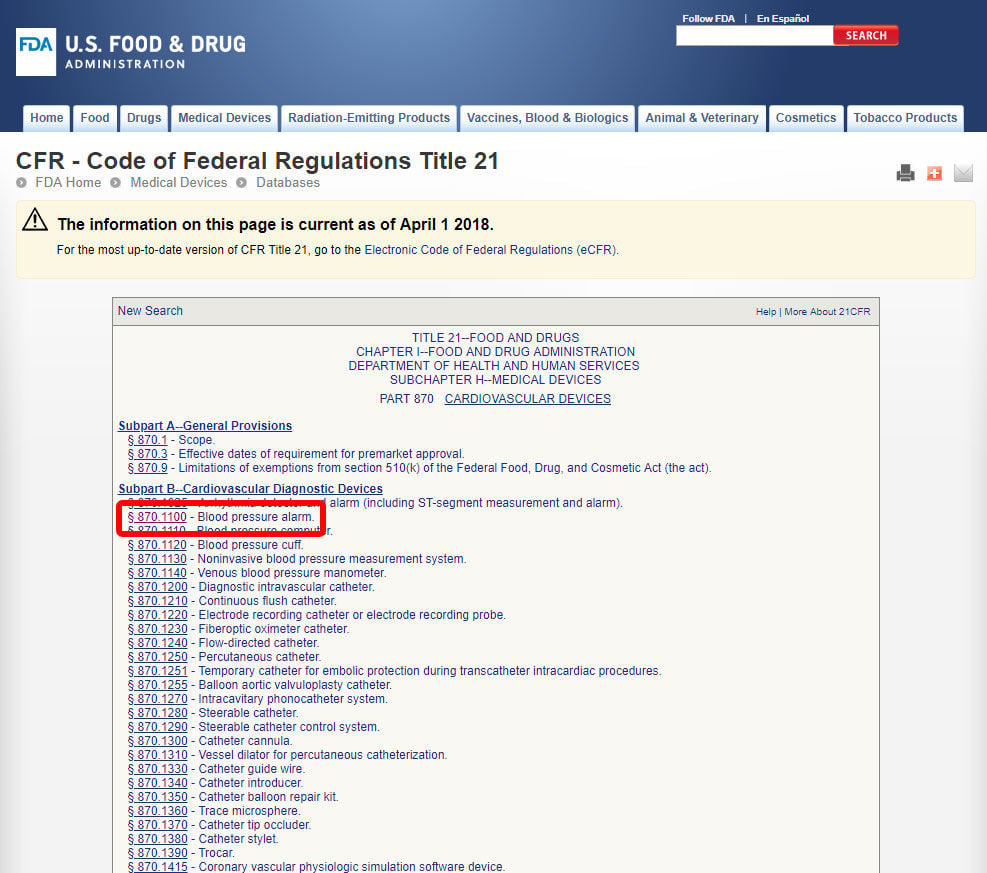
Clique no código do dispositivo e abra as Diretrizes. A classificação do dispositivo consta da secção b).,
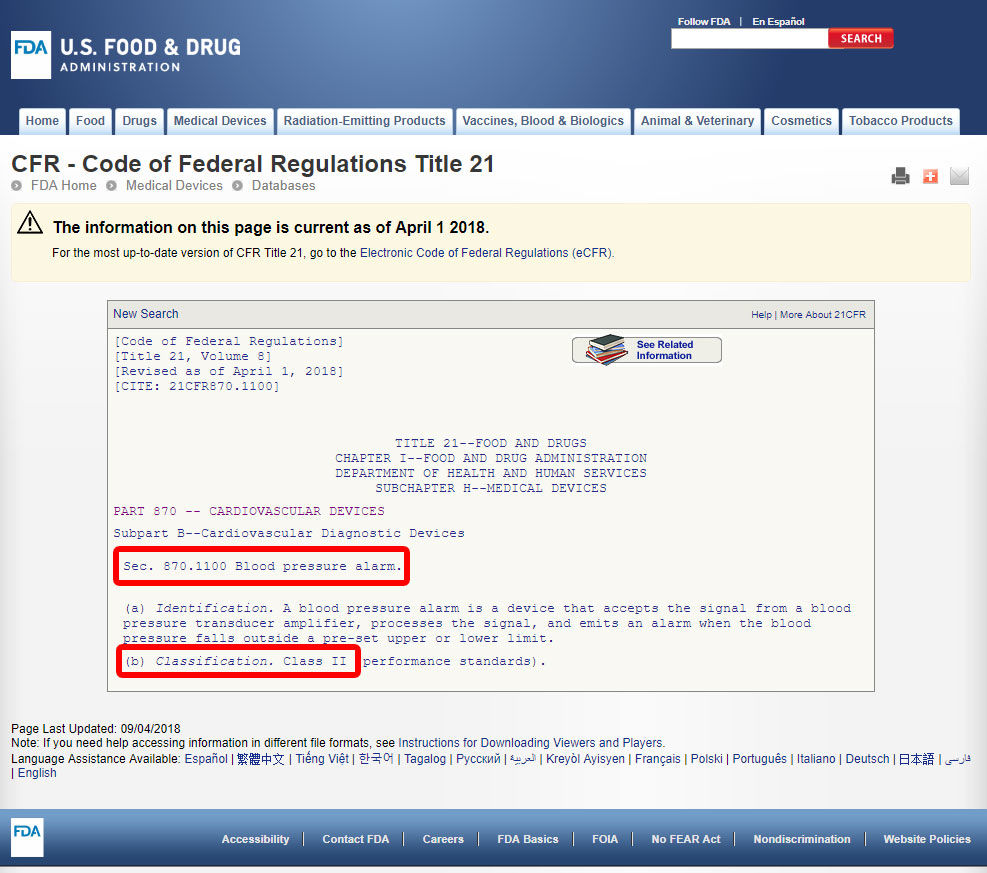
Se o seu dispositivo não possui um equivalente entre 1.700 dispositivos classificados pela FDA, é mais provável que um dispositivo inovador, sem um substancial equivalente e classificados como Classe III.
a Compreensão FDA Dispositivo Médico Classes
As diferenças entre dispositivos médicos classificados como Classe I, II ou III pelo FDA é, principalmente, o risco, a quantidade de contato com o paciente e seus sistemas internos, e se um dispositivo é fundamental para a manutenção da vida.,
além destes fatores, a FDA considera equivalência substancial ao determinar como um dispositivo é classificado. Se o seu dispositivo for de baixo risco e contactar minimamente o doente, é provável que se qualifique para a classe I e para um processo simplificado de aprovação do mercado. Os dispositivos das classes II e III devem demonstrar a segurança através de equivalência material, de um depósito de 510(k) ou do processo de aprovação do pré-mercado.ao saber como o seu dispositivo é classificado, pode racionalizar o seu caminho para a aprovação do mercado através da compreensão dos processos e documentos que provavelmente serão exigidos pela FDA., Se a sua organização está sujeita a um requisito de classe II ou classe III 510(k) ou PMA, este conhecimento pode ajudá-lo a alocar os recursos adequados com antecedência e planejar para um arquivo bem sucedido.
