Vilka är skillnaderna i FDA Medical Device klasser?
den amerikanska FDA reglerar alla medicintekniska produkter som marknadsförs i USA, som är grupperade i tre breda klasser. Alla medicintekniska produkter som godkänts av FDA klassificeras som antingen klass I, II eller III beroende på enhetens risk, invasivitet och påverkan på patientens övergripande hälsa. Men var är linjerna ritade mellan var och en av dessa tre klasser, och varför?,
de amerikanska FDA: s klassificeringsriktlinjer kan vara mycket förvirrande för tillverkare av medicintekniska produkter med begränsad exponering för systemet. Det finns en enorm skillnad i den optimala vägen till marknaden för tillverkare beroende på hur enheten är grupperad. Anordningar av klass i omfattas av betydligt färre lagstadgade krav än anordningar av klass II eller III.
genom att förstå skillnaderna i klasserna FDA medical device kan du förstå hur enheten grupperas., Med denna kunskap i hand kan tillverkare av medicintekniska produkter i förmarknadsstadierna bättre förbereda och fördela de resurser som behövs för regleringsgodkännande.
skillnader mellan FDA Medical device Classes
FDA har klassificerat över 1 700 olika typer av medicintekniska produkter. Enheterna är organiserade i Code of Federal Regulations (CFR) enligt 16 specialiteter, såsom kardiovaskulära eller hematologiska enheter., Klassificera din medicinska enhet enligt en av de 16 specialiteter är det första steget att förstå om du tillverkar en klass I, II eller III medicinsk enhet.
Efter att ha klassificerat en enhet enligt specialitet, instruerar FDA tillverkarna att fortsätta till förhandsmeddelande med kunskap om huruvida deras enhet är undantagen eller inte. Medicintekniska produkter av klass i, den minst riskfyllda och invasiva kategorin, är undantagna från anmälningsprocesserna före marknaden. Särskilda anordningar av klass II är också undantagna från förhandsgodkännande.,
alla enheter som regleras av FDA är dock föremål för nuvarande Good Manufacturing Practice (cGMP) krav för registrering, märkning och kvalitet. Men hur vet du om din enhet är klass i eller II, och om du är skyldig att genomgå premarket anmälan? 
Klass 1
Den AMERIKANSKA läkemedelsmyndigheten FDA definierar Klass jag enheter enheter som inte är avsedda för användning i att stödja eller bevara liv eller av väsentlig betydelse för att förebygga försämring av människors hälsa, och de får inte utgöra en potentiell orimlig risk för sjukdom eller skada.,”
dessa enheter är den vanligaste klassen av enheter som regleras av FDA, som utgör 47 procent av godkända enheter på marknaden.
klass i-enheter har minimal kontakt med patienter och låg inverkan på patientens övergripande hälsa. I allmänhet kommer Klass I-enheter inte i kontakt med patientens inre organ, centrala nervsystemet eller hjärt-kärlsystemet. Dessa enheter är föremål för minst antal lagstadgade krav.,
exempel på Klass I-enheter:
- elektrisk tandborste
- Tongue Depressor
- Oxygen Mask
- återanvändbar kirurgisk skalpell
- bandage
- sjukhusbäddar
att föra klass i medicintekniska produkter till marknaden
klass i-enheter är det snabbaste och enklaste att få ut på marknaden eftersom de presenterar den lägsta risken för patienten och är sällan kritisk till livsuppehållande Vård. Majoriteten av klass i-enheter är undantagna från FDA-krav för premarket Notification (510k) och premarket Approval (PMA).,
Klass I-enheter är inte undantagna från FDA: s allmänna kontroller, en serie kommandon som gäller för medicintekniska produkter av klass I, II och III. Bestämmelserna i denna lag gäller äktenskapsbrott, felaktighet, enhetsregistrering, register och god tillverkningssed. Tillverkare av medicintekniska produkter som faller i klass A är fortfarande skyldiga att genomföra ett kvalitetsledningssystem och följa standarder för att säkerställa en kvalitetsprodukt.,
relaterad läsning: skillnaden mellan premarket Notification 510 (k) och premarket Approval
klass 2
klass II medicintekniska produkter är mer komplicerade än klass i-enheter och utgör en högre riskkategori eftersom de är mer benägna att komma i långvarig kontakt med en patient. Detta kan innefatta enheter som kommer i kontakt med patientens kardiovaskulära system eller inre organ och diagnostiska verktyg.,
FDA definierar klass II-enheter som ”enheter för vilka allmänna kontroller är otillräckliga för att ge rimlig säkerhet och effektivitet hos enheten.,est Kit
att föra klass II medicintekniska produkter till marknaden
kontrollerna varierar beroende på enheten, men enligt FDA, kan inkludera:
- enhetens prestanda
- eftermarknadsövervakning
- patientregister
- särskilda märkningskrav
- premarket data requirements
- riktlinjer
patientregister
majoriteten av klass II-enheter är FDA godkända för marknaden genom premarket notification, eller 510(k) process.,
Klass II-enheter är föremål för samma allmänna kontroller som nämns ovan, men FDA definierar dem som ”enheter för vilka allmänna kontroller är otillräckliga för att ge rimlig säkerhet och effektivitet hos enheten.”Av den anledningen är klass II-enheter också föremål för särskilda kontroller. Dessa föreskrifter beror på enheten och kan innehålla speciella märkningskrav, patientregister och prestandastandarder.
de flesta klass II-enheter kommer till marknaden med hjälp av premarket Notification (510k) – processen., 510 (k) är en komplex applikation till FDA, vilket visar att en enhet är säker och effektiv genom att visa att enheten motsvarar en annan enhet som finns på marknaden.
denna process innebär att visa ”betydande ekvivalens” till en annan enhet som är känd i FDA parlance som ”predikatet.”Detta betyder inte att enheterna måste vara identiska, men de kräver betydande likheter vid användning, design, material, märkning, standarder och andra egenskaper.,
FDA släppte en undantagslista i början av 2018 som undantar över 800 generiska medicintekniska produkter av klass I och II från 510(k) – processen. Om du har en generisk klass II-medicinsk enhet kan du upptäcka om den är undantagen från en 510(k) arkivering genom att söka i FDA-produktklassificeringsdatabasen.
relaterad läsning: 5 Skäl översyn FDA 510(k) är ett bra drag.
klass 3
FDA definierar klass III-enheter som produkter som ”vanligtvis upprätthåller eller stöder livet, implanteras eller utgör en potentiell orimlig risk för sjukdom eller skada.,”
bara 10 procent av de enheter som regleras av amerikanska FDA faller i klass III. denna klassificering är i allmänhet utökad till permanenta implantat, smarta medicinska enheter och livsuppehållande system.
medan Klass III i allmänhet är reserverad för de mest innovativa och avancerade medicintekniska produkterna finns det andra produkter som kan falla i klass III av olika skäl., Vissa enheter som ursprungligen kategoriseras som klass II kan stötas upp till klass III om tillverkaren inte kan visa väsentlig ekvivalens med ett predikat (befintlig produkt) under arkiveringsprocessen för PMA (510k).,
exempel på medicintekniska produkter av klass III:
- bröstimplantat
- Pacemakers
- defibrillatorer
- högfrekventa ventilatorer
- cochleaimplantat
- provtagningsskärmar för fosterblod
- implanterade proteser
att föra medicintekniska produkter av klass III till marknaden
klass III-enheter är föremål för alla FDA general Controls och FDA premarket approval (PMA) process., FDA skriver: ”på grund av risknivån i samband med klass III-enheter har FDA bestämt att allmänna och speciella kontroller ensamma är otillräckliga för att säkerställa säkerheten och effektiviteten hos Klass III-enheter.”
PMA är den mest intensiva typen av enhetsmarknadsföring som krävs av FDA. Vissa FDA klass III-enheter är undantagna och kan kvalificera sig för en 510(k) arkivering, men majoriteten förväntas få Premarket godkännande.,
PMA-processen kräver en noggrann studie av en medicinsk produkt för att bevisa säkerhet och effektivitet genom utveckling av en datadriven nytta / riskprofil. PMA-processen omfattar i allmänhet kliniska prövningar och betydande tid och resurser för tillräcklig datainsamling. De enda undantagen från PMA-processen inom Klass III är anordningar med en väsentlig motsvarighet. Du kan avgöra om en klass III-enhet kan marknadsföras med en 510 (k) genom att söka i FDA Premarket Approval (PMA) – databasen och 510(k) premarket Notification database.,
hur du bestämmer din klass
det första steget mot att klassificera din medicinska enhet är att navigera i FDA: s Klassificeringsregler, listan över 16 kategorier för medicintekniska produkter enligt medicinsk specialisering.
som ett exempel visar vi dig stegen för att identifiera klassificeringen av ett blodtryckslarm. Enheten klassificeras under kategori 870: kardiovaskulära enheter.,
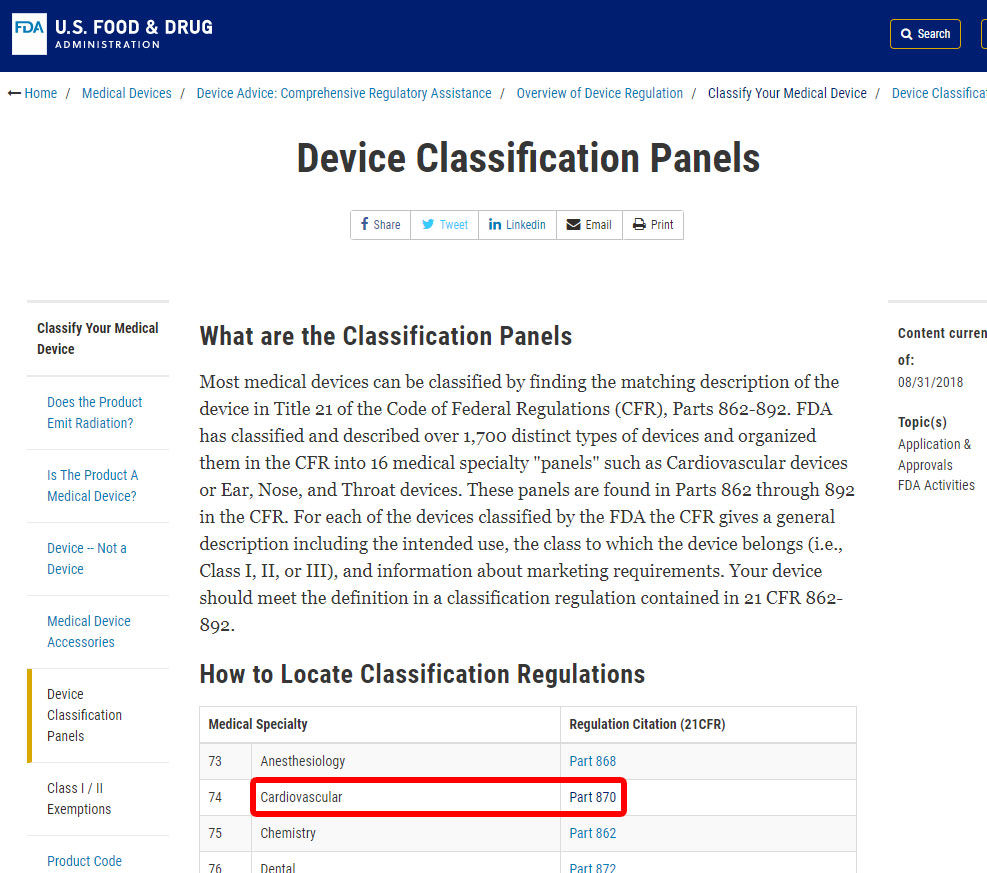
När du har hittat relevant medicinsk specialitet klickar du på kategorin och navigerar i listan över enheter tills du hittar en motsvarande och tillhörande enhetskod.
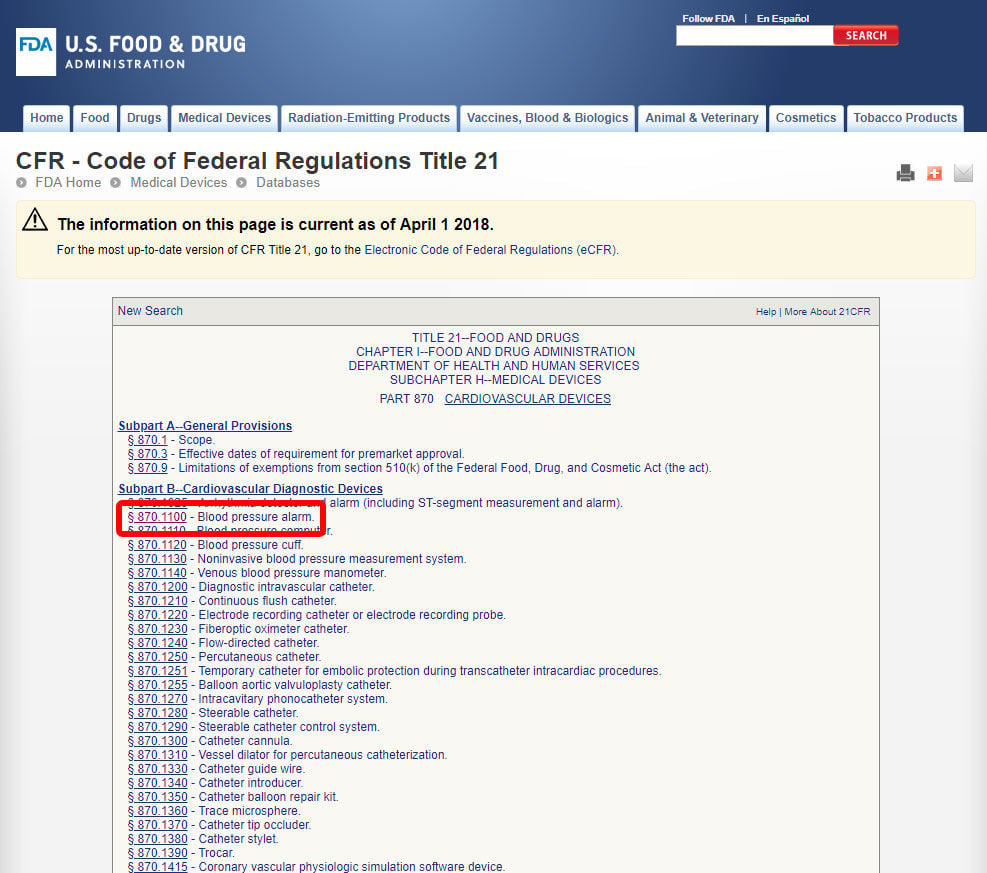
klicka på enhetskoden och öppna riktlinjerna. Produktklassificeringen anges i avsnitt b.,
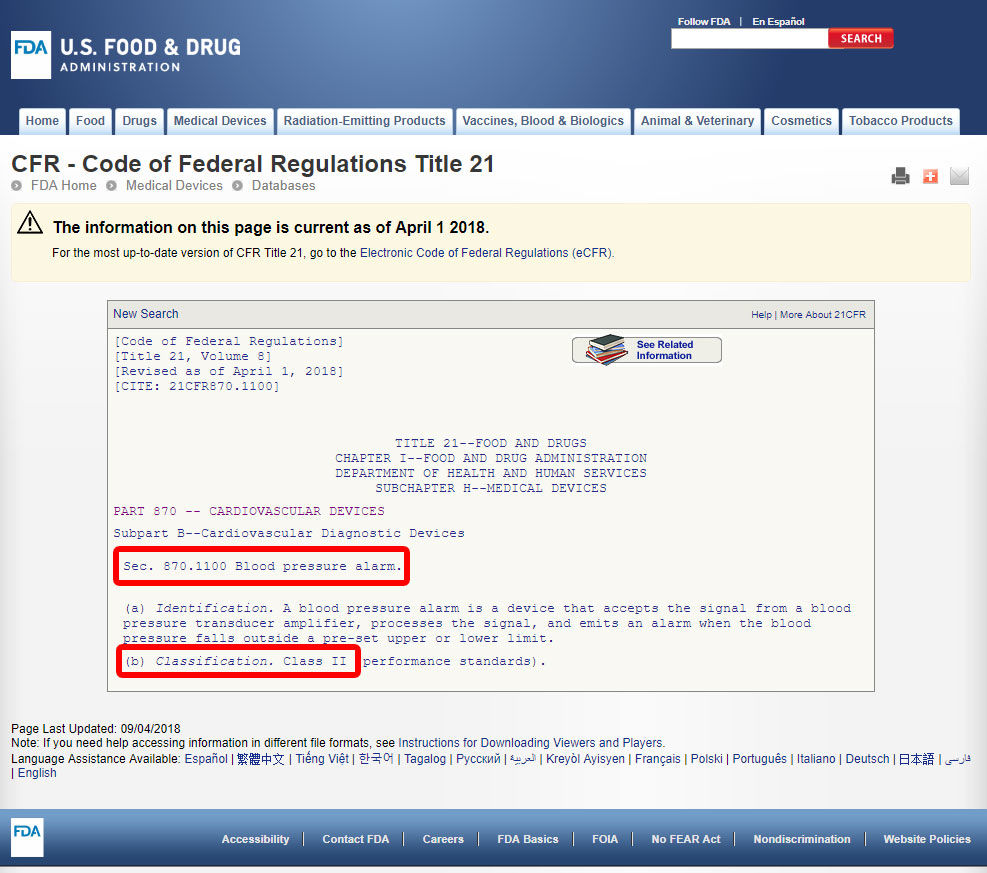
om din enhet saknar en listad motsvarighet bland de 1700 enheter som klassificerats av FDA, är det troligtvis en innovativ enhet utan väsentlig motsvarighet och skulle klassificeras som klass III.
förstå klasserna för medicintekniska produkter för FDA
skillnaderna mellan medicintekniska produkter klassificerade som klass I, II eller III av FDA är oftast risk, mängden kontakt med en patient och deras interna system, och om en enhet avgörande för att upprätthålla livet.,
utöver dessa faktorer anser FDA betydande ekvivalens vid bestämning av hur en enhet klassificeras. Om din enhet är lågrisk och minimalt kontaktar patienten, kommer du sannolikt att kvalificera dig för klass i och en strömlinjeformad marknadsgodkännandeprocess. Klass II-och klass III-produkter måste visa säkerhet genom materiell likvärdighet, en 510 k-ansökan eller förfarandet för godkännande av marknaden.
genom att veta hur din enhet klassificeras kan du effektivisera din väg till marknadsgodkännande genom att förstå de processer och dokument som sannolikt kommer att krävas av FDA., Om din organisation är föremål för ett Klass II-eller klass III 510(k) eller PMA-krav, kan denna kunskap hjälpa dig att fördela lämpliga resurser i förväg och planera för en lyckad arkivering.
