Nauwkeurige gendeletie en vervanging met behulp van het CRISPR/Cas9-systeem in menselijke cellen
Hier tonen we aan dat twee gidsrna ‘ s in combinatie met Cas9 efficiënt DNA-deleties van maximaal 10 kb genereren in menselijke cellen in een proces waarbij herstel van de deletie grotendeels wordt bereikt door nauwkeurige eindverbinding. Bovendien verstrekken wij gegevens die aantonen dat het systeem CRISPR/Cas9 grote genomic fragmenten in aanwezigheid van een lineaire homologe reparatiedonor kan vervangen.,
de bacteriële geclusterde korte palindromische herhalingen/CRISPR—geassocieerde (CRISPR / Cas) loci coderen RNA-geleide immuunsystemen die cellen beschermen tegen binnenvallende virussen en plasmiden (1, 2). In Streptococcus pyogenes, gebruiken de systemen type II CRISPR / Cas een RNA-geleide endonuclease (rgen), Cas9, om plaats-specifieke splitsing van de opeenvolgingen van doeldna te katalyseren., Het richten van Cas9 aan specifieke genomic plaatsen wordt bemiddeld door een opeenvolging van 20 nucleotidegids binnen een geassocieerd CRISPR RNA (crRNA) en vereist trans-activerende crRNA (tracrRNA) die crRNA in complex Cas9 (3) rekruteert. Erkenning van splitsingsplaatsen wordt bepaald door crRNA-DNA base pairing en een protospacer-aangrenzend motief (PAM), een drie nucleotide sequentie (NGG) naast elkaar geplaatst om het complementaire gebied van DNA (4)., Het is opmerkelijk dat één enkel gidsrna (gRNA) dat de complexe tracrrna-crRNA nabootst Cas9 aan gerichte genomic plaatsen kan rekruteren en dubbelstrengelde onderbrekingen (dsbs) in DNA (5) kan produceren. De systemen CRISPR / Cas9 zijn aangepast voor plaats-specifiek genoom het uitgeven in diverse celtypes en organismen (6-12).
genoombewerking met CRISPR / Cas9 wordt geïnitieerd met de introductie van een DSB op een gerichte genomische locus met behulp van het RNA-geprogrammeerde RGEN. Dit wordt gevolgd door de reparatie van de DSB door middel van homology-directed repair (HDR) of nonhomologe end-joining (NHEJ)., In aanwezigheid van een homologe reparatiedonor, kan het systeem CRISPR/Cas9 worden gebruikt om nauwkeurige en gedefinieerde wijzigingen en inserties bij een gerichte plaats door het HDR-proces te produceren. Bij afwezigheid van een homologe reparatiedonor, worden de enige DSBs die door CRISPR/Cas9 worden geproduceerd hersteld door foutgevoelige NHEJ, die in insertie of schrapping (indel) veranderingen resulteert. Indel-mutaties in coderende exonen kunnen voortijdige stop-codons of frame-shift-mutaties introduceren, waardoor de overeenkomstige eiwitten worden geïnactiveerd., De veranderingen Indel die van het herstellen van één enkel DSB worden gegenereerd kunnen niet nuttig zijn in experimenten gericht op het karakteriseren van de functionele domeinen van eiwit-codeert genen of voor inactivering van genomic elementen zoals intergenic of intronic regelgevende opeenvolgingen of noncoding genen van RNA. De schrappingen van het fragment van DNA in doelloci zouden een weg verstrekken om deze functionele elementen te bestuderen. Te dien einde, zijn veelvoudige DSBs geà ntroduceerd om schrappingen in Drosophila (12, 13), zebrafish (14), en menselijke cellen (8) te produceren, zij het met lage efficiency., De gerichte genomic deletions van DNA zijn ook bereikt gebruikend nuclease van de zinkvinger (ZFN) of transcriptie activator-als effector nuclease (TALEN) in menselijke cellen (15-17). De efficiëntie van deze benaderingen is echter over het algemeen laag. Bovendien blijven Zfns en TALENs enigszins moeilijk en duur om in de cellulaire context te ontwerpen, te ontwikkelen en empirisch te testen.
hier onderzochten we het ontstaan van fragment deleties in menselijke cellen, gekatalyseerd door het CRISPR/Cas9 systeem. We tonen aan dat 2 gRNAs in combinatie met Cas9 efficiënt DNA-verwijderingen tot 10 kb kunnen creëren., Van belang, vonden we dat de reparatie van dit verwijderingsproces grotendeels wordt bereikt door nauwkeurige einde toetreding. Bovendien lijkt gerichte schrapping met CRISPR/Cas9 onafhankelijk te zijn van de transcriptionele status van de beoogde locus. Ten slotte tonen wij aan dat het systeem CRISPR/ Cas9 kan worden gebruikt om grote genomic fragmenten in aanwezigheid van een lineaire homologe reparatiedonor te vervangen.
materiaal en methoden
Plasmideconstructie
de basis-H1-promotor werd versterkt uit pLVTHM-plasmide (Addgene, #12247, Cambridge, MA)., Oligonucleotiden die de gewijzigde promotor H1 en de ruggengraat van gewenste Grna-opeenvolgingen met twee plaatsen BsaI bevatten werden samengesteld (PAN faciliteit, Stanford Universiteit). De resulterende full-length producten werden versterkt door PCR en gekloond in de puc19 vector. Het ampicillin-gen (amp) en de H1-promotor in de puc19-vector bevatten bsai-restrictie-enzymplaatsen; deze werden gemuteerd (het AMP-gen werd veranderd van G1601C, wat de aminozuursequentie niet verandert; de H1-promotor werd veranderd van GAGACC in GAGGACC) om de bsai-plaatsen te elimineren., Het protocol voor gRNA het klonen wordt gepresenteerd in het aanvullende materiaal. Alle sequenties van doelgebieden worden weergegeven in aanvullende tabel S1.
celcultuur
HEK 293T, SK-Hep1 en HeLa cellen werden gekweekt in Dulbecco “s modified Eagle” s medium (DMEM) aangevuld met 10% foetaal runderserum (FBS) (Hyclone, Logan, UT) en penicilline/streptomycine (pen/strep) (Invitrogen, Carlsbad, CA). PC3 cellen werden gekweekt in rpmi-1640 medium aangevuld met 10% FBS en pen/strep., Voor tumornecrosefactor α (TNF-α)-stimulatie werden 293T-cellen behandeld met aangegeven concentraties TNF-α(R&D-systemen, Minneapolis, MN). In een bevochtigde incubator werden de cellen op 37°C en 5% CO2 gehouden.
gerichte DNA-deletie
HEK 293T-cellen werden gezaaid in 12-putplaten met een dichtheid van 100.000 cellen per put. Na 24 uur werden de cellen tijdelijk getransfecteerd met 1 µg Cas9 plasmide (Addgene, # 41815), 0,5 µg gRNA T1, en 0,5 µg gRNA T2 plasmiden met behulp van Lipofectamine 2000 (Invitrogen) volgens de protocollen van de fabrikant., Genomisch DNA werd 48 uur na transfectie geëxtraheerd met behulp van QuickExtract DNA-Extractieoplossing (Epicentre Biotechnologies, Madison, WI). Gemeenschappelijke PCR werd geleid om het gerichte gebied te versterken gebruikend primers flankerend de gerichte gebieden. Wild-type en afgeknotte genomic fragmenten werden opgelost door gelelektroforese. Real-time PCR (RT-PCR) werd uitgevoerd om het percentage van schrapping te kwantificeren gebruikend primers over de verbinding of binnen het schrappingsgebied. De vergelijkende cq-methode werd gebruikt om het expressieniveau van het doelgebied ten opzichte van een referentiegebied (ACTB locus) te berekenen., Het percentage van schrapping in het doelgebied werd verder berekend door de verhouding van doelcellen ten opzichte van controlecellen. Alle primer sequenties zijn vermeld in aanvullende tabel S2.
Doelsequencing
cellen werden twee dagen na transfectie geoogst en het genomische DNA werd geëxtraheerd met behulp van QuickExtract DNA-Extractieoplossing (Epicenter biotechnologieën). PCR werd geleid om het het richten gebied met genomic DNA uit de cellen wordt afgeleid te vergroten, en amplicons werden diep gerangschikt door MiSeq persoonlijke Sequencer (Illumina, San Diego, CA).,
Targeted DNA replacement
The linear donor was generated by PCR from pGl3-GFP-SV40pA plasmid, created by replacing the Renilla gene with the GFP gene in pRL-TK (Promega, Madison, WI). The primer sequences used for PCR were:
CCL2-donor-F
A*C*AGCAGCCAGAGGAACCGAGAGGCTGAGACTAACCCAGAAACATCCAATGCTTTTACGCGTCCTAGCG
CCL2-donor-R
C*A*AAAATATATTTATTTGGTGTAATAG TTACAAAATATTCATTTCCACAACCACCTGGATCCTTATCGA
The underlined regions indicate the termini of analogous oligonucleotides with 50 bp of CCL2 homology., De donorsequenties worden weergegeven in het aanvullende materiaal. De twee 5 ‘ – meeste verbindingen zijn fosforothioaat (aangeduid met sterretjes). Cellen in 6-Wells platen werden tijdelijk getransfecteerd met 2,0 µg Cas9 plasmide, 0,8 µg gRNA T1 plasmide, 0,8 µg gRNA T2 plasmide en 0,4 µg lineaire donor met behulp van Lipofectamine 2000 (Invitrogen). 48 uur na transfectie werden de cellen behandeld met 1 ng/mL TNF-α gedurende 24 uur, waarna GFP-positieve cellen werden gesorteerd.
Luciferasebepaling
voor de luciferasebepaling werden HEK 293T-cellen gezaaid in 96-wells-platen met een dichtheid van 5000 cellen per put., Na 24 uur werden de cellen tijdelijk getransfecteerd met 5 ng PRL-TK Renilla luciferase reporter en 100 ng luciferase reporter met cytomegalovirus (CMV), SV40 (Simian virus 40), of basispromotor. Na 48 uur werd de activiteit van luciferase gemeten met het dual luciferase reporter assay system (Promega).
Western blot
eiwitten werden gescheiden door natriumdodecylsulfaat—PAGE (SDS-PAGE) en overgebracht naar nitrocellulosemembranen. De membranen werden geblokkeerd met 5% vetvrije melk en geïncubeerd met GFP antilichaam (CST, #2555S, Danvers, MA)., Het antigeen-antilichaamcomplex werd gedetecteerd met versterkte chemiluminescentiereagentia.
resultaten en discussie
we hebben het bacteriële type II CRISPR/ Cas9 systeem aangepast om genomisch DNA in menselijke cellen te mutageniseren. De menselijke codon-geoptimaliseerde versie van S. pyogenes Cas9 proteïne die een C-terminus SV40 kernlokalisatiesignaal draagt werd uitgedrukt gebruikend een eerder beschreven systeem (6). Om Cas9 splitsing aan de gewenste opeenvolging te leiden, drukten wij crRNA-tracrRNA fusietranscripten uit, hierna als gids RNAs (gRNAs) wordt bedoeld, van een gewijzigde menselijke polymerase III promotor van H1., Het 3 ‘ eind van de promotor H1 werd gewijzigd om transcriptie van gRNAs toe te staan die met om het even welke nucleotide begint. Slechts beperkt door de eis dat het 20 BP crRNA-doel door de Pam-opeenvolging wordt gevolgd, NGG (waar n om het even welke nucleotide is), kan deze benadering in principe worden gebruikt om om het even welke genomische plaats te richten die de vorm N20NGG heeft. Om het klonen van de Grna expressievector te vergemakkelijken, gebruikten we een type IIS restrictie enzym, BsaI. Dit vereiste de synthese van een oligonucleotide van 24 BP die een gebied van complementariteit aan de doelplaats op DNA bevatten., Het eenvoudige en efficiënte protocol voor het klonen van de Grna-expressievector (figuur 1A) wordt in detail beschreven in het aanvullende materiaal.
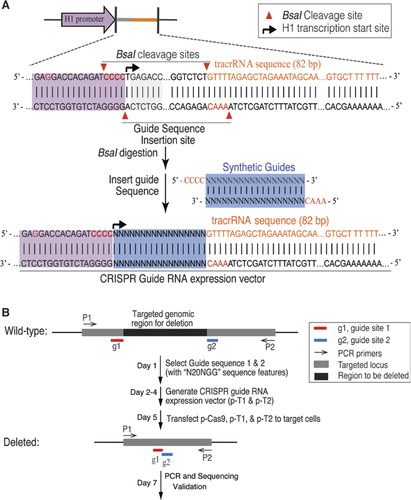
(A) ontwerp van de guide RNA (gRNA) expressie vector. De vector werd ontworpen om Grna-afschriften met synthetische Grna te produceren die aan trans-activerend RNA/CRISPR RNA (tracrRNA) worden gesmolten., De H1-promotor werd aangepast om het interne type IIS-restrictie-enzym bsai-locatie te elimineren door gagacc in de H1-promotor te veranderen in GAGGACC. Een bsai-plaats werd geà ntroduceerd om het klonen plaatsen voor Grna en tracrRNA fusies tot stand te brengen door toevoeging van synthetische oligonucleotide duplexen met compatibele overhangen. Het 3 ‘eind van de promotor H1 werd gewijzigd om CCACAGATCCCC te zijn om transcriptie van gRNAs met om het even welke nucleotide aan het 5’ eind te vergemakkelijken. (B) de stappen van gerichte genschrapping met CRISPR/Cas9.,
Voor het verwijderen van een groot segment van genomisch DNA gebruikten we een paar gRNAs tegen de beoogde locus (figuur 1B). Twee doelplaatsen met het patroon N20NGG werden geselecteerd op de grens van het doelgebied. De efficiency van gerichte schrapping geleid door diverse combinaties van Grna paren werden bepaald door PCR analyses gebruikend primers flankerend de gerichte gebieden. Wild-type en afgeknotte genomic fragmenten werden opgelost door gelelektroforese. Om PCR-versterkingsvooroordeel te vermijden, werd de percentageschrapping gekwantificeerd door RT-PCR gebruikend één inleidingspaar., De primers werden ontworpen over de schrapping junction (een primer buiten het schrappingsgebied, de andere primer binnen het schrappingsgebied) of binnen de schrappingsgebieden (beide primers vinden binnen het schrappingsgebied). Aldus, wordt slechts één enkele band met het inleidingspaar voor zowel de gerichte cellen als controlecellen versterkt. We berekenden procent schrapping door het vergelijken van de relatieve hoeveelheid PCR-producten (doelcellen versus controlecellen) versterkt door hetzelfde primerpaar. Doelgerichte verwijderingen werden verder geverifieerd door sequencing.,
om te beoordelen hoe gRNA-paren later herstel en generatie van verwijderingen zouden kunnen beïnvloeden, ontwierpen we eerst sets Grna ‘ s gericht tegen de menselijke CDC42 genomische locus en gescheiden door afstanden variërend van ongeveer 200 tot 10.000 bp (figuur 2A en aanvullende tabel S1). Wij beoordeelden toen de capaciteit van elk gRNA paar om schrappingen in menselijke HEK 293T cellen in aanwezigheid van Cas9 te produceren. Robuuste efficiëntieverbeteringen van op NHEJ gebaseerde verwijderingen (tot 68%) werden bevestigd door qPCR (figuur 2B–2D)., Zelfs voor schrapping van een 10 kb genomic gebied, verkregen we targeting tarieven van 16% tot 28%, afhankelijk van de gRNA paar. Dit RNA-gemedieerde bewerkingsproces was snel, met de eerste detecteerbare deletie verschijnen ongeveer 12 uur post-transfectie (aanvullende figuur S1). Het systeem was effectief in een verscheidenheid van celtypes, met inbegrip van: PC3, SK-Hep1, en HeLa cellen (aanvullend figuur S2).

(A) schematische weergave van de locaties van guide RNAs (gRNAs) gericht op de CDC42 locus. (B-D) de efficiëntie van gerichte schrapping met CRISPR/Cas9 werd bepaald door PCR in HEK 293T cellen. Primers buiten de verwachte deletie regio ‘ s werden gebruikt. Het percentage van schrapping werd gekwantificeerd door RT-PCR gebruikend inleidingen over de verbinding of binnen het schrappingsgebied. (E-F) de efficiëntie en precisie van gerichte schrapping in CDC42 werden bevestigd door (E) Sanger-sequencing en (F) high-throughput-sequencing analyses. Het PCR-product dat alleen het deletieamplicon bevat, werd verrijkt voor sequencing.,
verwijderingen werden verder bevestigd door het sequencen van PCR-producten over de verwachte splitsingsplaatsen. Het rangschikken van Sanger toonde schrappingverbindingen voortkwam uit de nauwkeurige afbinding van de botte – beëindigde dsbs die door Cas9 wordt gecreeerd; elke DSB kwam precies 3 bp vóór de Pam-opeenvolging voor (figuur 2E en aanvullend cijfer S3). We gebruikten ook diepe sequencing van schrapping amplicons om de nauwkeurigheid van schrapping efficiëntie te beoordelen; in ongeveer 80% van de reads, de gerichte DSBs werden perfect gerepareerd (figuur 2F).,
we hebben deze bevindingen samengevat door gRNA-paren te testen die bedoeld waren om fragmenten te verwijderen uit een genomische locus die het microRNA mir-21 gen in HEK 293T-cellen bevat. Twee gRNAs werden ontworpen om de grenzen van de MIR-21 haarspeld (aanvullende figuur S4) te richten. De deletie-efficiëntie was 38% na transfectie met de twee gRNAs en Cas9, zoals gemeten met een PCR-test (aanvullend figuur S4B). Het diepe rangschikken bevestigde de schrapping kwam precies voor zoals verwacht (aanvullend cijfer S4C).,
om te onderzoeken of het CRISPR / Cas9-gemedieerde gen deletiesysteem wordt beïnvloed door de transcriptionele staat van de beoogde genen, werd het gen dat het chemokine (C-C-motief) ligand 2 (CCL2) codeert, gericht. CCL2 is een klein cytokine behorend tot de CC chemokine familie; het ccl2 gen is een doel van NF-kB signaleren. We selecteerden willekeurig acht doelplaatsen in de 5′ en 3 ‘ uiteinden van de ccl2 gen locus (figuur 3A). We bereikten robuuste en efficiënte deletie van verschillende regio ‘ s van het gen met behulp van Cas9 en verschillende paren gRNAs in HEK 293T cellen (figuur 3B–3D).,

(A) schematische weergave van de locaties van guide RNAs (gRNAs) gericht op de ccl2 locus. (B-D) de efficiëntie van gerichte schrapping van CCL2 met CRISPR/Cas9 in HEK 293T-cellen. (E) ccl2 mRNA-spiegels werden bepaald na toevoeging van tumornecrosefactor α (TNF-α) door kwantitatieve RT-PCR-analyses in HEK 293T-cellen. De gegevens werden getoond met de middelen ± sem in drievoud., F) de efficiëntie van gerichte deletie van CCL2 met CRISPR/Cas9 na behandeling met TNF-α gedurende 24 uur in HEK 293T-cellen. G) het niveau van luciferase-activiteit van cytomegalovirus (CMV), of SV40, of een basispromotor in HEK 293T-cellen. De gegevens werden getoond met de middelen ± sem in drievoud. H) efficiëntie van gerichte deletie van een luciferasegen gecontroleerd door CMV, of SV40, of een basispromotor in HEK 293T-cellen. (B–D,F,H) het percentage van schrapping werd gekwantificeerd door RT-PCR gebruikend primers over de kruising of binnen het schrappingsgebied.,
expressie van het ccl2-gen werd dramatisch geïnduceerd door TNF-α (tot ∼300-voudige toename) in HEK 293T-cellen (figuur 3E), wat een goed model biedt om te onderzoeken of de CRISPR/ Cas9-gemedieerde gerichte genoombewerking wordt beïnvloed door transcriptionele activiteit. Van belang is dat de efficiëntie van gerichte deletie van de ccl2-genlocus niet werd beïnvloed door de behandeling van cellen met TNF-α(figuur 3F), wat suggereert dat transcriptie de CRISPR/Cas9-gemedieerde deletie niet veranderde., Om dit resultaat verder te bevestigen, richtten we ons op een exogeen verslaggeversgen dat wordt aangedreven door verschillende promotors met verschillende sterktes, waar de verschillende transcriptionele activiteiten kunnen worden geëvalueerd met behulp van een luciferaseanalyse (figuur 3G). PCR-analyses toonden vergelijkbare efficiëntie aan in gerichte deleties in HEK 293T-cellen na co-transfectie van het verslaggeversgen samen met Cas9-en gRNA-paren (figuur 3H). Dit resultaat geeft aan dat nhej-gemedieerde reparatie kan optreden ondanks het optreden van verschillende graden van transcriptionele activiteit.,
DSBs kan HDR stimuleren om zeer nauwkeurige vervanging van het beschadigde gebied door een homologe donor mogelijk te maken. Om gerichte genomic vervanging van DNA te verkrijgen, introduceerden wij een paar gRNAs, Cas9, en een lineaire donor met homologie aan het gerichte gebied in cellen (figuur 4A). De lineaire donor werd verkregen door PCR-versterking met primers met een 50 bp homologe sequentie. Dezelfde donor werd met succes ingebracht met behulp van een op ZFN gebaseerd HDR-reparatiesysteem (18)., Om de haalbaarheid van CRISPR/ Cas9-gemedieerde vervanging door HDR te testen, richtten we ons op de ccl2 locus met een paar gRNAs (#39 en #1854 getoond in Figuur 3) en een donor die de verbeterde groene fluorescente eiwit (EGFP) codeersequentie en SV40 poly(A) plaats draagt (figuur 4A; sequenties en posities worden gepresenteerd in het aanvullende materiaal). Gebruikend dit systeem, was ongeveer 0.5% van gerichte cellen EGFP-positief, terwijl slechts 0.023% EGFP-positief in schijntransfectiecellen was (enkel getransfecteerd met donor), die aan controlecellen gelijkaardig was (0.021%, zonder transfectie)., De EGFP-positieve cellen werden dan gesorteerd door cytometry stroom. Plaats-specifieke integratie werd bevestigd door PCR met behulp van twee paren primers flankeren zowel de homologe armen en de hele vervangen regio. Zoals getoond in Figuur 4B, merkten wij het verwachte vervangen gebied dat de volledige lengtesegfp opeenvolging en de homologe wapens bevat (het rangschikkende resultaat van Sanger dat in het aanvullende materiaal wordt getoond). Het endogene wild-type allel werd ook gedetecteerd (figuur 4B), wat erop wijst dat niet alle allelen het doelwit zijn., Verder selecteerden we enkele klonen uit de EGFP-positieve cellen en vonden dat alle klonen (6 van de 6 onderzochte) de verwachte integratie hadden (figuur 4C), maar het endogene wild-type allel werd ook gedetecteerd in drie van de klonen (figuur 4C), wat suggereert dat er slechts één allel was gericht in die klonen. Expressie van EGFP-eiwit in gerichte cellen (EGFP-positief gesorteerde cellen) werd up-gereguleerd op TNF-α behandeling zoals geëvalueerd door Western blot en fluorescentie geactiveerd cel Sorteren (FACS) (Figuur 4, D en E)., Deze resultaten toonden aan dat het systeem CRISPR/Cas9 kan worden gebruikt om gen/domeinvervangingen met hoog rendement en nauwkeurigheid tot stand te brengen.

(A) schematische schema ‘ s die de procedure van gerichte genvervanging met behulp van CRISPR/Cas9 in menselijke cellen weergeven., Om de werkzaamheid van gerichte genvervanging te testen, werden guide RNAs (gRNAs) ontworpen om het aangegeven gebied (gekenmerkt door plaatsen 1 en 2) van het ccl2 gen te verwijderen en om het verwijderde gebied te vervangen door de egfp-polyA cassettedonor met armen met korte gebieden van homologie. De gerichte plaatsen van plaats 1 en plaats 2 zijn #39 en #1854 binnen het ccl2 gen getoond in Figuur 3. De homologe opeenvolgingen (50 bp) zijn enkel stroomopwaarts en stroomafwaarts van de schrappingplaatsen. De posities en sequenties in detail worden weergegeven in het aanvullende materiaal., (B) de efficiëntie van gerichte genvervanging met CRISPR/Cas9 werd bepaald door PCR in HEK 293T-cellen. De inleidingen die de verbindingen tussen CCL2 en EGFP overspannen werden gebruikt voor PCR-versterking. C) PCR-test voor de gerichte genvervanging van de enkele klonen. (D,E) expressie van EGFP-eiwit op TNF-α-toevoeging in HEK 293T-cellen werd bepaald door (D) Western blot en (e) FACS.
Hier beschrijven we een eenvoudige en efficiënte aanpak voor Gen deletie met behulp van het CRISPR/Cas9 systeem., We toonden aan dat de introductie van dit systeem in menselijke HEK 293T-cellen, en andere menselijke celtypen, geleid heeft tot deleties van fragmenten tot 10 kb met efficiënties variërend van 11% tot 68%, afhankelijk van de beoogde sequentie. De capaciteit om genomic segmenten efficiënt en nauwkeurig te schrappen zal de studie van functionele genomic elementen in menselijke cellen vergemakkelijken. Deze benadering kan potentieel worden gebruikt om om het even welke genomic loci te richten.
Er was bezorgdheid over de specificiteit van het CRISPR / Cas9-systeem (19-21)., Om ongewenste fenotypen als gevolg van mutaties buiten het doel uit te sluiten, stellen wij voor dat voor elk doelgebied ten minste twee verschillende paren gRNAs worden gebruikt. In onze studie was de behoefte aan meerdere Grna-paren geen belangrijke beperking, gezien de eenvoud en zeer efficiëntie van dit systeem. Het is opmerkelijk dat verschillende paren gRNAs gericht op dezelfde regio werkte met een hoge efficiëntie (figuren 2 en 4). Een andere benadering om ongewenste mutaties te voorkomen is het gebruik van de dubbele nickase methode (22, 23)., We hebben ook met succes de dubbele nickase methode toegepast om schrapping van genomisch DNA te genereren, maar de efficiëntie was aanzienlijk lager.
Het is bekend dat het herstel van DNA-DSBs grotendeels wordt gemedieerd door foutgevoelig NHEJ, waarbij de twee uiteinden worden verwerkt en aan elkaar geligerd op een manier die vaak gepaard gaat met nucleotide-inserties en-deleties. Dergelijke foutgevoelige eindverbinding werd waargenomen bij de reparatie van DSBs gemaakt door ZFNs of TALENs. Daarentegen was de reparatie van DSBs die door Cas9 en twee gRNAs werd gegenereerd, zeer nauwkeurig., Onze resultaten suggereren dat de pauzes direct afliggen zonder eindverwerking, het onthullen van een eerder niet-gewaardeerd voordeel van de nhej-route. Het mechanisme dat resulteert in de precieze ligaties moet nog worden bepaald. Een mogelijkheid is dat gerichte schrapping met behulp van Cas9 en twee gRNAs resulteert in een verbinding die niet wordt herkend door een van de originele gRNAs. Wij analyseerden ook de efficiency in het produceren van indelveranderingen voor het individuele Grna en gRNA paar (#39 en #224 gRNA in Figuur 3A) door sanger het rangschikken van PCR amplicons (TA het klonen)., Van belang, merkten we op dat de efficiëntie van het genereren van Indel mutaties voor single gRNA vrij laag was (9,5%, 2 van 21 klonen voor #39 gRNA; 5%, 1 van 20 klonen voor #224 gRNA). Het gRNA-paar genereerde echter een hoog rendement van Indel-mutaties (50%, 10 van de 20 klonen voor # 39 en # 224), wat vergelijkbaar was met de test met qPCR (52%, figuur 3B). Wij stellen voor dat een enkele gRNA resulteert vaak in een stomp einde van de splitsing site, die precies zal worden gerepareerd door NHEJ. Aldus, is de efficiency van het produceren van verandering veel lager met gebruik van één enkele gRNA dan een paar gRNAs.,
Dankbetuigingen
Dit werk werd ondersteund door subsidies van het National Institute of Health (nr. DP1CA174421) en de W. M. Keck foundation aan C.-Z. C, en de National Natural Science Foundation of China (No.81101481) en Shanghai Medisch Talent Training Program (No. XYQ2011048) aan S. L. H. deze paper is onderworpen aan het NIH Public Access Policy.
concurrerende belangen
De auteurs verklaren geen concurrerende belangen.
aanvullende gegevens
om de aanvullende gegevens bij dit document te bekijken, kunt u terecht op de website van het tijdschrift op: www.,future-science.com/doi/suppl/10.2144/000114196